lucianoemi
Usuario (Argentina)
Las malformaciones de Chiari son un grupo de anomalías caracterizadas por presentar un desplazamiento caudal del tronco cerebral y cerebelo. Existen 6 tipos: -Tipo 0: Descenso del bulbo pero no de las amígdalas. -Tipo I: Descenso de las amígdalas cerebelosas por debajo del foramen magno. Se asocia a siringomielia cervical o cervicotoracica. Predomina en adultos, alrededor de los 40 años. -Tipo II: Descenso de las amígdalas y vermis cerebelosos, IV° ventrículo y bulbo por debajo del foramen magno. Se asocia a mielomeningocele. Predomina en neonatos -Tipo 1,5: Similar al tipo II pero sin disrafia espinal asociada. -Tipo III: Descenso del cerebelo en un encefalocele occipital. -Tipo IV: Hipoplasia cerebelosa. Siringomielia: Se llama siringomielia a la cavidad quística intramedular, tapizada (hidromielia) o no por epéndimo y rodeada por tejido glial. Si bien no es una anomalía del desarrollo se la incluye en este capitulo por su fuerte asociación con las malformaciones de Chiari. También, puede ser secundaria a traumatismos, tumores y aracnoiditis espinales. Existen casos en que no se encuentra patología asociada. En ocasiones, la cavidad se extiende al bulbo (siringobulbia). Predomina en los adultos jóvenes. La fisiopatología del Chiari y la siringomielia es compleja. Probablemente exista un trastorno en la circulación de LCR entre el cráneo y la columna (Chiari I) o dentro del espacio subaracnoideo espinal (trauma, tumores y aracnoiditis espinales) que desciende las amígdalas y/o fuerza al LCR dentro de la médula a través del óbex. Existen varias teorías: Teoría hidrodinámica de Gardner: el drenaje del IV° ventrículo se encuentra ocluído, razón por la cual el efecto de "martillo de agua" de la onda de pulso normal del LCR ocasiona el descenso amigdalino a través del foramen magno y la entrada de liquido dentro de la médula por el óbex. Teoría de la disociación cráneo-espinal de Williams: la malformación de Chiari, durante el ciclo cardiaco, por un mecanismo valvulado unidireccional permite la entrada de LCR dentro del cráneo pero no su salida, generando un gradiente cráneo-espinal que ocasiona el descenso amigdalino y la penetracion de liquido dentro de la médula por el óbex. Teoría de Ball y Dayan: cuando existe una obstrucción crónica local en la columna (disco, meningioma espinal o fibrosis) el LCR no circula libremente por el espacio subaracnoideo espinal y penetra dentro de la médula por los espacios de Virchow-Robin. Teoría de Klekamp: las obstrucciones a la circulación del LCR impiden el drenaje normal de liquido extracelular intramedular al espacio subaracnoideo y éste se acumularía dentro de la médula, formando cavidades. La clínica del Chiari varía según la edad de presentación: Neonatos: estridor laríngeo y disfagia que progresan hacia una severa disfunción bulbar en días o semanas. Infancia y adultos: cefalea suboccipital (aumenta con la tos), cuadriparesia espástica, nistagmo, ataxia cerebelosa, diplopía, disartria, disfonía y disfagia de evolución lenta. LA MALFORMACIÓN DE CHIARI EN UN ADULTO ES UNA ENTIDAD QUE DEBE SER SOSPECHADA ANTE LA PRESENCIA DE UNA CONSTELACIÓN DE SÍNTOMAS APARENTEMENTE INCONEXOS ENTRE SÍ. La clínica de la siringomielia refleja el compromiso intramedular. Los síntomas aparecen espontáneamente, después de una maniobra de Valsalva o de un trauma: -Síndrome de compresión "centromedular" total o parcial. -Disociación termoalgesica en forma de banda suspendida (solo en el 50%). -Dolor referido de origen medular. -Atrofia muscular en las manos. -Escoliosis. El diagnostico de Chiari y siringomielia se efectúa con las IRM que muestran: El descenso amigdalino a través del foramen magno, considerado anormal cuando es mayor de 5 mm. La cavidad quistica intramedular, hipointensa en las imágenes ponderadas en T1 e hiperintensa en las imágenes ponderadas en T2 por su contenido de LCR, cuyas paredes no refuerzan luego de la administración intravenosa de contraste. El tratamiento es quirúrgico y sus objetivos son eliminar la compresión y normalizar la circulación del LCR. En los casos con síntomas progresivos se recomienda el tratamiento precoz. La cirugía consiste en una o varias de las siguientes técnicas: Craniectomía de la fosa posterior con apertura del foramen magno y laminectomia de C1, C2 y/o C3, para eliminar la compresión de las amígdalas cerebelosas sobre el tronco cerebral. Plástica meníngea para ampliar y reconstruir la cisterna magna y restablecer la circulación normal de LCR. Drenaje de la cavidad siringomielica mediante un catéter hacia el espacio subaracnoideo espinal o hacia las cavidades pleural o peritoneal. Remoción de la causa (discopatias - tumores) cuando sea posible. En los neonatos la cirugía fracasa en 1/3 de los casos, falleciendo por falla respiratoria, debido a la lesión irreversible del tronco cerebral. En los niños y adultos la respuesta a la cirugía es mucho más favorable, deteniéndose la progresión de los síntomas en la gran mayoría de los casos.
La espasticidad es un trastorno motor del sistema nervioso en el que algunos músculos se mantienen permanentemente contraídos. Dicha contracción provoca la rigidez y acortamiento de los músculos e interfiere sus distintos movimientos y funciones: deambulación, manipulación, equilibrio, habla, deglución, etc. La espasticidad está causada normalmente por daños en las zonas del cerebro o de la médula espinal que controlan la musculatura voluntaria. Suele aparecer asociada a traumatismos del cerebro o de la médula espinal, esclerosis múltiple, parálisis cerebral, hipoxia o ictus cerebral, Enfermedad de Tay-Sachs, algunos desórdenes metabólicos como la adrenoleucodistrofia o la fenilcetonuria. Cursa habitualmente con hipertonía (aumento del tono muscular), calambres (rápidas contracciones sin movimiento notable), espasmos (contracciones con movimiento) e hiperreflexia de tendones profundos (reflejos exagerados). El grado de espasticidad varía desde una leve rigidez muscular hasta graves, dolorosos e incontrolables espasmos musculares. Tratamiento no farmacológico: Rehabilitación: fisioterapia de estiramiento y movilidad muscular,crioterapia, terapia ocupacional, medidas ortopédicas (para la corrección y prevención de contracturas musculares o dolores articulares mediante el empleo de dispositivos externos conocidos como ortesis), estimulación eléctrica (transcutánea), y como última solución la cirugía empleada para liberar tendones, contracturas musculares o miofasciales (fibrotomia gradual), o canales nerviosos (rizotomía). Tratamiento farmacológico: El tratamiento farmacológico consiste en: Medicamentos cuya acción se dirige al sistema nervioso central: baclofeno, benzodiazepinas (diazepam y clonazepam), diazepam, tizanidina, gabapentina, clonidina así como el cannabis y sus derivados. Medicamentos cuya acción se dirige al sistema nervioso periférico: son dantroleno -retirado del mercado español- e inyecciones de fenol y toxina botulínica A. En España están disponibles solamente el baclofeno, el diazepan y la tizanidina como medicamentos con autorización para el tratamiento de la espasticidad. "ESPASTICIDAD: La espasticidad es un trastorno motor del sistema nervioso en el que algunos músculos se mantienen permanentemente contraídos. Dicha contracción provoca la rigidez y acortamiento de los músculos e interfiere sus distintos movimientos y funciones: deambulación, manipulación, equilibrio, habla, deglución, etc. La espasticidad está causada normalmente por daños en las zonas del cerebro o de la médula espinal que controlan la musculatura voluntaria. Suele aparecer asociada a traumatismos del cerebro o de la médula espinal, esclerosis múltiple, parálisis cerebral, hipoxia o ictus cerebral, Enfermedad de Tay-Sachs, algunos desórdenes metabólicos como la adrenoleucodistrofia o la fenilcetonuria. Cursa habitualmente con hipertonía (aumento del tono muscular), calambres (rápidas contracciones sin movimiento notable), espasmos (contracciones con movimiento) e hiperreflexia de tendones profundos (reflejos exagerados). El grado de espasticidad varía desde una leve rigidez muscular hasta graves, dolorosos e incontrolables espasmos musculares. Tratamiento no farmacológico: Rehabilitación: fisioterapia de estiramiento y movilidad muscular,crioterapia, terapia ocupacional, medidas ortopédicas (para la corrección y prevención de contracturas musculares o dolores articulares mediante el empleo de dispositivos externos conocidos como ortesis), estimulación eléctrica (transcutánea), y como última solución la cirugía empleada para liberar tendones, contracturas musculares o miofasciales (fibrotomia gradual), o canales nerviosos (rizotomía). Tratamiento farmacológico: El tratamiento farmacológico consiste en: Medicamentos cuya acción se dirige al sistema nervioso central: baclofeno, benzodiazepinas (diazepam y clonazepam), diazepam, tizanidina, gabapentina, clonidina así como el cannabis y sus derivados. Medicamentos cuya acción se dirige al sistema nervioso periférico: son dantroleno -retirado del mercado español- e inyecciones de fenol y toxina botulínica A. En España están disponibles solamente el baclofeno, el diazepan y la tizanidina como medicamentos con autorización para el tratamiento de la espasticidad."
La progeria de Hutchinson-Gilford es un síndrome extremadamente raro caracterizado por un envejecimiento prematuro de inicio postnatal. Las características clínicas y radiológicas principales incluyen alopecia, piel fina, ausencia de grasa subcutánea, rigidez articular y osteolisis. La inteligencia no está afectada. La muerte prematura se produce por arterioesclerosis o enfermedad cerebro-vascular. Descripción El nombre del síndrome deriva de la palabra griega geras, que significa viejo, y fue acuñado por Gilford en 1904. El síndrome de envejecimiento prematuro, también llamado síndrome de Hutchinson-Gilford o progeria, es un cuadro caracterizado por una aceleración en el proceso natural de envejecimiento, que se produce en estos individuos en edades tempranas de la vida. Este proceso de envejecimiento se produce entre 5 y 10 veces más rápido de lo habitual, por lo que estos pacientes se ven afectados desde la infancia. Son niños sin retraso mental, con baja altura. La esperanza de vida de estos pacientes es corta, nunca supera los 30 años, con un promedio entre los 14 a 15 años. Su muerte ocurre generalmente en la adolescencia, la mayoría de las veces por enfermedades cardiovasculares. Causas e incidencia Actualmente se desconocen con exactitud las causas que aceleran el proceso de envejecimiento, aunque hay estudios que parecen indicar que esta enfermedad podría ser causada por un gen mutante autosómico dominante que haría inestable al núcleo de las células del organismo acelerando el proceso de muerte celular. Lamin A es el nombre del gen recientemente identificado como el causante de algunos tipos de progeria. La función del producto de este gen es desconocido, pero se piensa que estaría involucrado en la supresión del fenotipo de envejecimiento. Al encontrarse alterado, este gen no cumpliría bien su función supresora, fallando en último caso el proceso de frenado del envejecimiento. La progeria sería en este caso una enfermedad genética, lo que no significa que tenga que ser necesariamente una enfermedad hereditaria. La mayoría de los casos se producen por mutaciones esporádicas, es decir mutaciones que se producen en el gen del individuo afectado, y no transmitidas por sus progenitores. La incidencia de la progeria clásica ha sido estimada en 1/8 000 000 de recién nacidos vivos. No se ha evidenciado preferencia por ningún sexo en particular, pero se han comunicado muchos más pacientes de raza blanca (97% de los pacientes afectados). Síntomas La progeria ha llegado a ser una enfermedad de conocimiento público, debido a que sus síntomas, que incluyen adelgazamiento y manchas de la piel, reabsorción de la masa ósea, pérdida de cabello y arteriosclerosis, se asemejan bastante al envejecimiento humano normal. La progeria produce un rápido envejecimiento de los niños, comenzando con una deficiencia en el crecimiento durante el primer año de vida que tiene como resultado cuerpos desproporcionadamente pequeños en relación con el tamaño de sus cabezas. Los niños son delgados y presentan calvicie, cara alargada y arrugada y piel de apariencia envejecida. Los niños con progeria desarrollan aterosclerosis temprana. La esperanza de vida para estos niños, si bien es muy variable, nunca supera los 30 años. La piel es delgada y con escaso tejido celular subcutáneo, las uñas son quebradizas, curvadas, amarillentas e incluso pueden faltar en algunos dedos. La muerte prematura se produce por arteriosclerosis o enfermedad cerebro-vascular y falta de medro. Estos son los principales síntomas característicos de la progeria: Deficiencia en el crecimiento durante el primer año de vida Cara alargada y arrugada Mentón retraído Ojos saltones y nariz en forma de pico Calvicie Pérdida de las pestañas y las cejas Estatura baja Cabeza grande para el tamaño de la cara (macrocefalia) La fontanela (punto blando) permanece abierta Mandíbula pequeña (micrognatia) Piel seca, descamativa y delgada Pecho estrecho Abdomen abultado Huesos deformes Enfermedades degenerativas, como la artritis, propias de la vejez Rango de movimiento limitado Retardo en la formación o ausencia de los dientes Subir al menú Diagnóstico El diagnóstico de la progeria es fundamentalmente clínico y se plantea en niños que presentan los signos iniciales de enfermedad entre el primer y segundo año de vida y que manifiestan las características fundamentales de la enfermedad. No existe ninguna prueba que pueda establecer por si misma el diagnóstico definitivo de progeria, pero este puede apoyarse en algunas pruebas bioquímicas, histológicas y radiológicas. Alteraciones radiológicas: cara y cráneo fontanelas permeables, huesos wormianos con fracturas, hipoplasia malar y mandibular, dientes apiñados; hipoplasia clavicular en el tórax, costillas ahusadas; largos identaciones en los huesos, corticales delgadas, metáfisis anchas, coxa valga, coxa plana, genu valgum; en las falanges acro-osteolisis progresiva de las falanges distales; y otros signos como osteoporosis, escoliosis, cuerpos vertebrales en forma de boca de pescado, luxación de cadera, falta de unión de fracturas y pérdida de tejidos blandos. Rayos X de tórax (vista anteroposterior). Osteólisis total de ambas clavículas y de las primeras costillas. Osteólisis parcial de las segundas y terceras costillas 1- Rayos X de tórax (vista anteroposterior). Osteólisis total de ambas clavículas y de las primeras costillas. Osteólisis parcial de las segundas y terceras costillas. Rayos X de columna lumbosacra (vista lateral). Persistencia de la escotadura en cara anterior de las vértebras dorsales (vértebras en fishmouth). Espondilolistesis de la quinta vértebra lumbar sobre la primera sacra. 2- Rayos X de columna lumbosacra (vista lateral). Persistencia de la escotadura en cara anterior de las vértebras dorsales (vértebras en fishmouth). Espondilolistesis de la quinta vértebra lumbar sobre la primera sacra. Rayos X de ambos pies (vista anteroposterior). Osteólisis parcial de falanges distales de ambos pies. 3- Rayos X de ambos pies (vista anteroposterior). Osteólisis parcial de falanges distales de ambos pies. Rayos X de pelvis ósea (vista anteroposterior). Coxa valga bilateral. 4- Rayos X de pelvis ósea (vista anteroposterior). Coxa valga bilateral. Características histopatológicas: incluyen alteraciones de la piel como áreas de piel normal que alterna con zonas de esclerodermia, la dermis es delgada y presenta zonas de desorganización de fibras colágenas y áreas de hialinización. A nivel del tejido subcutáneo se evidencia pérdida de tejido graso, múltiples anomalías en el hueso, tipo: osteolisis, osteoporosis, displasia esquelética, necrosis avascular de la cabeza femoral, luxación de cadera y fracturas sin resolución. A nivel cardiovascular se encuentran placas ateromatosas, presentes en todos los vasos, pero más prominentes en las arterias coronarias, la aorta y las mesentéricas. Otro hallazgo son las calcificaciones de las válvulas aórtica y mitral, así como signos de fibrosis, isquemia e infarto del miocardio. Alteraciones bioquímicas: el hallazgo más constante en estos pacientes ha sido el aumento en la excreción de ácido hialurónico en la orina, evento que no se ha podido relacionar con trastornos endocrinos ni evento dismetabólico alguno.Subir al menú Complicaciones Debido a que la progeria induce envejecimiento precoz, el organismo de los afectados suele desarrollar las siguientes enfermedades: Aterosclerosis Arteriosclerosis Artritis Artrosis Diabetes mellitas Embolias Hipercolesterolemia Hipertensión Osteoporosis Tratamiento Como se desconocen con exactitud las causas que desencadenan la aceleración de la velocidad de envejecimiento y muerte celular, no existe en la actualidad un tratamiento específico capaz de revertir dicha aceleración. Por lo tanto, tendremos que conformarnos con tratar individualmente cada síntoma o complicación que vaya surgiendo con el proceso natural de la enfermedad; intentando limitar al máximo posible su influencia en la vida de los pacientes. Según los investigadores que hallaron la mutación genética responsable del cuadro, este descubrimiento ofrece la clave para hallar en un futuro la curación, como así también conocer aún más el proceso de envejecimiento normal. Es importante que estos niños desarrollen una vida social lo más normal que se pueda, dentro de sus posibilidades, teniendo en cuenta que el desarrollo de la inteligencia no se ve afectado.
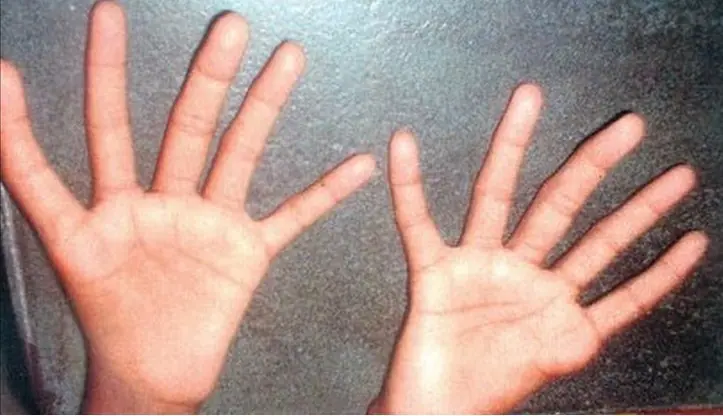
Es un raro trastorno que involucra anemia y ciertas deformidades esqueléticas y articulares. Causas La mayoría de los casos del síndrome de Aase ocurren sin una razón conocida y no se transmiten de padres a hijos (hereditarios). Sin embargo, se ha demostrado que algunos casos son hereditarios. Esta afección es similar a la anemia de Diamond-Blackfan y las dos enfermedades no se deben separar. Un fragmento faltante en el cromosoma 19 se encuentra en algunas personas con este tipo de anemia. La anemia en el síndrome de Aase es causada por el desarrollo insuficiente de la médula ósea, que es donde se forman las células sanguíneas. Síntomas Nudillos pequeños o ausentes Paladar hendido Disminución de los pliegues cutáneos en las articulaciones de los dedos de la mano Orejas deformes Párpados caídos Incapacidad para extender completamente las articulaciones desde el nacimiento (deformidad por contractura) Hombros estrechos Piel pálida Pulgares trifalángicos Pruebas y exámenes El médico llevará a cabo un examen físico. Los exámenes que se pueden hacer abarcan: Biopsia de médula ósea Conteo sanguíneo completo (CSC) Ecocardiografía Radiografías Tratamiento El tratamiento puede consistir en transfusiones de sangre durante el primer año de vida para tratar la anemia. Un medicamento esteroide llamado prednisona también se ha utilizado para tratar la anemia asociada con este síndrome; sin embargo, se debe usar únicamente después de revisar los beneficios y riesgos con un médico que tenga experiencia en el tratamiento de anemias. Si otro tratamiento falla, puede ser necesario realizar un trasplante de médula ósea. Expectativas (pronóstico) La anemia tiende a mejorar con la edad. Posibles complicaciones Las complicaciones relacionadas con la anemia abarcan: Fatiga Disminución del oxígeno en la sangre Debilidad Los problemas cardíacos pueden llevar a una variedad de complicaciones, dependiendo del defecto específico. Los casos severos de este síndrome han estado asociados con mortinatos o muerte prematura. Prevención Se recomienda la asesoría genética si usted tiene antecedentes familiares de este síndrome y desea quedar embarazada. Nombres alternativos Síndrome de Aase-Smith; Anemia hipoplásica/síndrome del pulgar trifalángico
La ataxia telangiectasia es una enfermedad rara que pertenece al grupo de los síndromes neurocutáneos, pues implica alteraciones tanto en la piel como en el sistema nervioso central. Se trata de una enfermedad genética causada por una mutación que afecta al gen ataxia telangiectasia (Atm), localizado en el brazo largo del cromosoma 11. Es una afección autosómica recesiva, por lo que ambos padres han tenido que aportar un gen defectuoso para que el niño acabe heredando la enfermedad. La ataxia telangiectasia se manifiesta en etapas tempranas de la infancia, aunque no suele ser visible en el momento del nacimiento. En general, los afectados muestran: Ataxia cerebelosa progresiva, lo que conlleva dificultades motoras, que se comienzan a notar entre el primer y el cuarto año de vida. Telangiectasias (vasos sanguíneos dilatados que ofrecen un aspecto similar a una araña vascular). Las telangiectasias aparecen en la nariz, las orejas, la parte interna de rodillas y codo y en la conjuntiva de los ojos. Inmunodeficiencia , de lo que se deriva una mayor vulnerabilidad para padecer distintas y recurrentes infecciones en las vías respiratorias . Envejecimiento prematuro. Hipersensibilidad a las radiaciones ionizantes. Más probabilidad de padecer tumores, con un riesgo de cáncer cuatro veces mayor con respecto a otra persona sin ataxia telangiectasia. Dificultad progresiva al hablar. Decoloración de ciertas zonas de la piel. Problemas metabólico-endocrinos como la diabetes. Crecimiento más lento de lo normal. Ausencia o displasia de la glándula timo (que participa en la respuesta inmunitaria del organismo). Movimientos oculares anormales (apraxia progresiva). El diagnóstico de la ataxia telangiectasia puede hacerse a través de una exploración física, pues el desarrollo de los afectados muestra algunas peculiaridades. No obstante, como hay distintos tipos de ataxias, para diagnosticar la ataxia telangiectasia se puede recurrir a pruebas genéticas que detecten si hay mutaciones en el gen implicado. También otros exámenes médicos, como una radiografía en el timo o una analítica para determinar si la alfafetoproteína está elevada y si están bajos los indicadores IgA, IgG y/o IgE, pueden ser concluyentes. En algunos casos, la ataxia telangiectasia se confunde con la parálisis cerebral, ya que aparecen primero las dificultades del movimiento, y hasta varios años más tarde, usualmente sobre los seis años de edad, las telangiectasias, que son las que confirman el diagnóstico. Hasta el momento no existe ningún tratamiento curativo para la ataxia telangiectasia. La terapéutica va destinada a controlar algunos de los síntomas propios de la enfermedad. Así, se suele indicar fisioterapia y ejercicio para fortalecer la musculatura y evitar contracturas y se aconseja también la fisioterapia torácica para favorecer el drenaje de bronquios y pulmones. Además, para controlar la extensión de las telangiectasias, conviene limitar la exposición al sol y hay que evitar, en la medida de lo posible, las radiaciones ionizantes de las radiografías. Para proteger frente a las reincidentes infecciones respiratorias, pueden establecerse distintos tratamientos profilácticos con vacunas, antibióticos e inmunoglobulinas. Los afectados por ataxia telangiectasia tienen una esperanza de vida limitada, que suele estar en torno a los 25 años, aunque hay casos que han doblado esa edad. Lo más usual es que hacia los 10 años las dificultades motoras hagan que el niño tenga que utilizar una silla de ruedas. La ataxia telangiectasia es hereditaria. La única forma de prevenir su transmisión de padres a hijos es mediante el diagnóstico genético preimplantacional, una técnica de reproducción asistida en la que se escogen los cromosomas libres de la enfermedad antes de ser implantados en el útero materno. Un hijo de ambos padres portadores de ataxia telangiectasia tiene un 25% de posibilidades de resultar afectado, un 50% de ser portador y un 25% de no estar afectado ni ser portador. La ataxia telangiectasia es detectable también en el primer trimestre de embarazo mediante la amniocentesis. Amniocentesis: Video Uno de cada 40.000-100.000 nacidos vivos padece ataxia telangiectasia. Las diferencias están en el grado de consanguinidad de cada comunidad
Síndrome de Brunner es la hipótesis de que un trastorno genético poco común causada por una mutación en el gen MAOA. Se caracteriza por una menor que el coeficiente intelectual promedio, comportamiento impulsivo problemáticas, trastornos del sueño y cambios de humor. Sólo se ha identificado en catorce machos de una misma familia. Causas Síndrome de Brunner es causada por una deficiencia de la monoamina oxidasa A, que conduce a un exceso de monoaminas en el cerebro, como la serotonina, la dopamina y la norepinefrina. En tanto en ratones y seres humanos, una mutación se encuentra en el octavo exón del gen MAO-A, que creó un disfuncional gen MAO-A. La función normal de la MAO-A, rompiendo las monoaminas, se interrumpe y monoaminas acumularse en el cerebro. Los ratones que carecían de un funcional gen MAO-A muestran niveles más altos de agresión, en comparación con ratones con un funcional gen MAO-A. Historia Síndrome de Brunner fue descrito en 1993 por HG Brunner et al en el descubrimiento de un defecto genético particular en los miembros masculinos de una gran familia holandesa. Brunner encontró que todos los miembros varones de la familia con este defecto hace reaccionar agresivamente cuando está enojado, temeroso, o frustrado. El defecto descubierto se encontró más tarde que es una mutación en el gen que codifica para la monoamina oxidasa A. Brunner dijo que un "MAO-Una deficiencia está asociada con un fenotipo de comportamiento reconocible que incluye la regulación alterada de la agresión impulsiva". Una carta publicada por Hebebrand Klug y criticó las conclusiones de Brunner para no usar los estrictos criterios del DSM. Sociedad y cultura Resultados de Brunner se han utilizado para argumentar que la genética, en lugar de los procesos de toma de decisiones, puede causar la actividad criminal. La evidencia que apoya la defensa genética proviene de los resultados tanto de Brunner y una serie de estudios sobre ratones. Para demostrar la correlación entre la deficiencia de MAO-A y la agresión en los tribunales, a menudo se sostiene que los individuos no pueden ser considerados responsables de sus genes, y por lo tanto, no deben ser considerados responsables de sus disposiciones y acciones resultantes.