
1) SÍNDROME DE TOURETTE
El síndrome de Tourette es un trastorno neuropsiquiátrico heredado con inicio en la infancia, caracterizado por múltiples tics físicos (motores) y vocales (fónicos). Estos tics característicamente aumentan y disminuyen; se pueden suprimir temporalmente, y son precedidos por un impulso premonitorio. El síndrome de Tourette se define como parte de un espectro de trastornos por tics, que incluye tics transitorios y crónicos.
El síndrome de Tourette se consideraba un raro y extraño síndrome, a menudo asociado con la exclamación de palabras obscenas o comentarios socialmente inapropiados y despectivos (coprolalia), pero este síntoma está sólo presente en una pequeña minoría de afectados. El síndrome de Tourette ya no es considerado una enfermedad rara, pero no siempre es correctamente diagnosticado porque la mayoría de los casos son leves y la severidad de los tics disminuyen en la mayoría de los niños a su paso por la adolescencia. Entre 0,4% y el 3,8% de los niños de 5 a 18 años pueden tener el síndrome de Tourette; la prevalencia de tics transitorios y crónicos en niños en edad escolar es alta, y los tics más comunes son parpadeo de ojos, toser, carraspear, olfatear y movimientos faciales. Un Tourette grave en la edad adulta es una rareza, y el síndrome de Tourette no afecta negativamente a la inteligencia o la esperanza de vida.
Por lo general, los síntomas del síndrome de Tourette se manifiestan en el individuo antes de los 18 años de edad. Puede afectar a personas de cualquier grupo étnico y de cualquier sexo, aunque los varones lo sufren unas 3 ó 4 veces más que las mujeres.
El curso natural de la enfermedad varía entre pacientes. A pesar de que los síntomas oscilan entre leves hasta muy severos, en la mayoría de los casos son moderados.

2) SÍNDROME DE MOEBIUS
El síndrome de Möbius o de Moebius es una enfermedad neurológica congénita extremadamente rara. Dos importantes nervios craneales, el 6º y el 7º, no están totalmente desarrollados, lo que causa parálisis facial y falta de movimiento en los ojos. Estos nervios controlan tanto el parpadeo y el movimiento lateral de los ojos como las múltiples expresiones de la cara. También pueden estar afectados otros puntos del sistema nervioso, entre ellos otros nervios cerebrales que controlan otras sensaciones y funciones.
Los efectos clínicos son múltiples, entre otros, dificultades iniciales para tragar que pueden llevar a déficit de desarrollo y los problemas que conllevan la falta de sonrisa, el babeo, y el habla y la pronunciación defectuosas. Las alteraciones observadas en los ojos consisten principalmente en estrabismo y limitación del movimiento; afortunadamente, son poco frecuentes la ulceración de la córnea y otros cuadros relacionados con el mal desempeño de los párpados. Los problemas dentales aparecen pronto y reflejan la incapacidad del niño para una higiene bucal apropiada después de las comidas y que la boca permanezca entreabierta.
3) SÍNDROME DE ASPERGER
El síndrome de Asperger o trastorno de Asperger es un conjunto de problemas mentales y conductuales que forma parte de los trastornos del espectro autista. Se encuadra dentro de los trastornos generalizados del desarrollo (CIE-10;Capítulo V; F84). La persona afectada muestra dificultades en la interacción social y en la comunicación de gravedad variable, así como actividades e intereses en áreas que suelen ser muy restringidas y en muchos casos estereotípicas.
Se diferencia del autismo infantil temprano descrito por Kanner y de otras formas menos específicas en que en el trastorno de Asperger no se observa retraso en el desarrollo del lenguaje, y no existe una perturbación clínicamente significativa en su adquisición. No hay retardo, por ejemplo en la edad en que aparecen las primeras palabras y frases, aunque pueden existir particularidades cualitativas (por ejemplo gramaticales) que llamen la atención, así como una preservación generalizada de la inteligencia. Aunque la edad de aparición y detección más frecuente se sitúa en la infancia temprana, muchas de las características del trastorno se hacen notorias en fases más tardías del desarrollo, cuando las habilidades de contacto social comienzan a desempeñar un papel más central en la vida de la persona.
4) SÍNDROME DE KAWASAKI
El síndrome de Kawasaki es una enfermedad multisistémica, idiopática, caracterizada por vasculitis que afecta a vasos de pequeño y mediano calibre, especialmente a arterias coronarias provocando clásicamente aneurismas en éstas, además, suele asociarse a algún síndrome mucocutáneo. Este síndrome es llamado así por el médico japonés Tomisaku Kawasaki que en la década de 1960 describió esta enfermedad recientemente reconocida en niños.
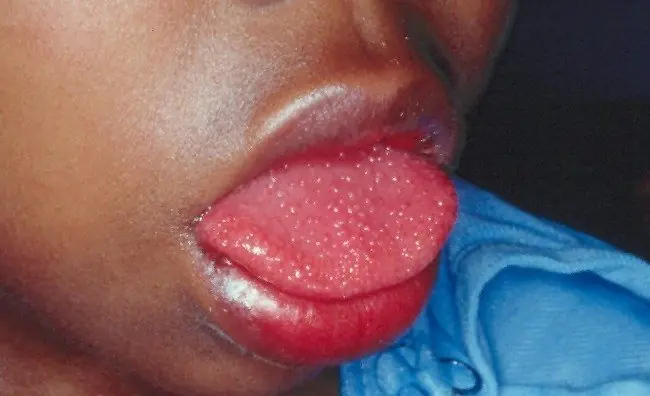
5) SÍNDROME DE TURNER
El síndrome Turner, síndrome Ullrich-Turner o monosomía X es una enfermedad genética caracterizada por la presencia de un solo cromosoma X. Tanto fenotípica como Genotípicamente son mujeres (por ausencia de cromosoma Y). Se trata de la única monosomía viable en humanos, dado que la carencia de cualquier otro cromosoma en la especie humana es letal. A las mujeres con síndrome de Turner les falta parte o todo un cromosoma X. En algunos casos se produce mosaicismo, es decir que la falta de cromosoma X no afecta a todas las células del cuerpo.
La ausencia de cromosoma Y determina el sexo femenino de todos los individuos afectados, y la ausencia del segundo cromosoma X determina la falta de desarrollo de los caracteres sexuales primarios y secundarios. Esto confiere a las mujeres que padecen el síndrome de Turner un aspecto infantil e infertilidad de por vida. Incide, aproximadamente, en 1 de cada 2.500 niñas.
Otros nombres alternativos son síndrome Bonnevie-Ullrich o disgenesia gonadal, monosomia X.
6) SÍNDROME DE MARFAN
El síndrome de Marfan es una enfermedad rara del tejido conectivo, que afecta a distintas estructuras, incluyendo esqueleto, pulmones, ojos, corazón y vasos sanguíneos.
Se caracteriza por un aumento inusual de la longitud de los miembros. Se cree que afecta a una de cada 5.000 personas y, a diferencia de otros problemas genéticos, no afecta negativamente a la inteligencia.

7) SÍNDROME DE EDWARDS
El síndrome de Edwards, también conocido como trisomía 18, es un tipo de aneuploidía humana que se caracteriza usualmente por la presencia de un cromosoma adicional completo en el par 18. También se puede presentar por la presencia parcial del cromosoma 18 (translocación desequilibrada) o por mosaicismo en las células fetales.
Fue originalmente descrita por John H. Edwards en la Universidad de Wisconsin, cuyos resultados fueron publicados y registrados en la literatura pediátrica y genética en el año 1960. Los estudios de genética molecular no han descrito con claridad las regiones puntuales que necesitan ser duplicadas para que se produzca el fenotipo característico del síndrome Edwards. Hasta el momento sólo se conocen dos regiones del brazo largo: 18q12-21 y 18q23.
8) SÍNDROME DE COTARD
El síndrome de Cotard, también llamado delirio de negación o delirio nihilista, es una enfermedad mental relacionada con la hipocondría. El afectado por el síndrome de Cotard cree estar muerto (tanto figurada como literalmente), estar sufriendo la putrefacción de los órganos o simplemente no existir. En algunos casos el paciente se cree incapaz de morir.
Recibe su nombre de Jules Cotard, neurólogo francés descubridor de este síndrome, al que denominó le délire de négation (delirio de negación), en una conferencia en París en 1880.
En dicha conferencia, Cotard describió el caso de una paciente -a la que dio el apodo de Mademoiselle X-, que negaba la existencia de Dios y el diablo, así como de diversas partes de su cuerpo y de la necesidad de nutrirse. Más adelante, creía que estaba eternamente condenada y que ya no podría morir de una muerte natural.
Los pacientes llegan a creer que sus órganos internos han paralizado toda función, que sus intestinos no funcionan, que su corazón no late, que no tienen nervios, ni sangre ni cerebro e incluso que se están pudriendo, llegando a presentar algunas alucinaciones olfativas que confirman su delirio (olores desagradables, como a carne en putrefacción), inclusive pueden llegar a decir que tienen gusanos deslizándose sobre su piel.
En sus formas más complejas el paciente llega a defender la idea de que en realidad él mismo está muerto e incluso que han fallecido personas allegadas a él. Junto con esta creencia de muerte el paciente mantiene una idea de inmortalidad, como si se hubiera convertido en un "alma en pena". Aunque es un delirio típico de las depresiones más graves (psicóticas o delirantes) se puede ver en otras enfermedades mentales severas (demencia con síntomas psicóticos, esquizofrenia, psicosis debidas a enfermedades médicas o a tóxicos).
Young y Leafhead describen un caso moderno de síndrome de Cotard en un paciente que sufrió daños cerebrales debido a un accidente de motocicleta:
Los síntomas [del paciente] se dieron en el contexto de sensaciones más generales de irrealidad y de estar muerto. En enero de 1990, después de recibir el alta en el hospital de Edimburgo, su madre lo llevó a Sudáfrica. Estaba convencido de que había sido llevado al infierno (lo que se confirmaba por el calor), y que había muerto de septicemia (que había sido un riesgo al principio de su recuperación), o quizá de sida (había leído una historia en The Scotsman acerca de alguien aquejado de sida que había muerto de septicemia), o de una sobredosis de una inyección contra la fiebre amarilla. Pensaba que se habían «apropiado del espíritu de mi madre para mostrarme el infierno», y que seguía dormido en Escocia.
El síndrome puede aparecer en el contexto de una enfermedad neurológica o mental y se asocia particularmente con la depresión y la desrealización.

9)SÍNDROME DE WEST
El síndrome de West (SW) o síndrome de los espasmos infantiles es una encefalopatía (alteración cerebral) epiléptica de la infancia, grave y poco frecuente, que debe su nombre a William James West (1793-1848), médico inglés que describió por primera vez el cuadro (presente en su propio hijo) en un artículo publicado por The Lancet en 1841. Se caracteriza típicamente por tres hallazgos: espasmos epilépticos, retraso del desarrollo psicomotor y electroencefalograma con un trazado característico de hipsarritmia, aunque uno de los tres puede no aparecer.
Los niños con SW suelen manifestar la enfermedad entre los 3 y 6 meses de edad, aunque en ocasiones esto ocurre hasta los dos años. El SW siempre genera algún grado de retraso global en el desarrollo infantil y, a pesar de que el conocimiento sobre él ha mejorado considerablemente, todavía hay casos en los que no se diagnostica a tiempo, ante todo cuando los síntomas son leves (las convulsiones se pueden confundir con cólicos o dolor abdominal) o debido a la falta de experiencia por parte del pediatra.
10) SÍNDROMDE DE GALINEAU (NARCOLEPSIA)
La narcolepsia es una enfermadad autoinmune cuya prevalencia en la población es muy baja. Se caracteriza por la presencia de accesos de somnolencia irresistible durante el día. Puede cursar con cataplejía (parálisis o debilidad extrema bilateral de un conjunto muscular), alucinaciones hipnagógicas (visiones fugaces en la transición vigilia-sueño) o hipnopómpicas (transición sueño-vigilia); incluso puede haber parálisis del sueño, e interrupción del sueño nocturno. De acuerdo con estudios epidemiológicos, la prevalencia de este trastorno neurológico del sueño en la población adulta se ubica entre un 0,02 y un 0,16%, afectando en forma similar a hombres y mujeres.
