yolachiqui
Usuario (Argentina)
Les dejo algunas imagenes para sacarle a alguno que a otro una linda sonrisa Bueno, espero que les hayan gustado! Saludos!

Aca estan .. http://www.todoelocio.net/images/el-mal-humor.jpg http://4.bp.blogspot.com/_t8P_QVW3XMo/SaHF4uXq1OI/AAAAAAAACJg/krDohrJ_H7s/S660/Humor%2Bgrafico.JPG http://www.humorgrafico.com.es/imagenes/river.JPG Espero les haya gustado.. Saludos Atte: Camila (Yolachiqui)
Taringueros..Taringueras...Poringueros,, Poringueras.. jajaj Nanna jodita lo primero. sis. Bueno qiero qe sepan qe nada.. hoy empezamos un nuevo año.. el año 2010, aunque para algunos noa haya sido mui lindo que digamos el 2009...epero qe este 2010 lo arranquen y lo terminen de lo mejor.. con toda la oda y bueno.. gracias .. gracias por esos post interesantes..gracias por hacerme reir..con esos comentarias taan graciosos qe todos hacen..gracias a los moderadores T! por fijarse qe la pagina ande bn qe nose sarpen por eliminar a los spameros a los post de los poringueros.. jaja na todo bn con los poringa. jajajaja y bueno..yo la verdad..es que teno ahce creo qe un par de años la cuenta.. soi re taringuera.. pero la posta es qe a principios de este añ o a fiens del ante año pasado eempeze a usar T! verdad.. y bueno me cope postee me hice NFU y yo estaba haci re emocion!! aajajaja Y bueno.. Taringa, es grosa. sabela. jajaja es atrapannte mal. jajaja y bueno garacias a los truqeros..osea timberos qe jgaron contra mi y no se dejaron ganar !! ¬¬ jajajaj igual tdo bn-.encximaa me matan! esaas imagenes qe ponen en los comentarios!! ajajaja bueno eso era omas. espeor se hayan tomado un tiempito haci chiqito para leer esto que vienne del corazon posta. Saludos a los metaleros, a los reggaetoneros, a los rolinga, a los cumbieros a los goticos.. y etc we jaja Besos espero empiezen el año con todo Gracias taringueros.. Gracias Taringa. PD: perdon por la ortografia.. no tengo faltas.. pero el teclado este es re feito Besoss!! Ahi les pongo unas de los tipi cos commentarios. jajaja y para cerrar la mejor qe ahora la van avver
El arte del Origami Para los occidentales nos resulta llamativo e ingenioso el arte de crear con papel todo tipo de figuras con determinados dobleces. Es una afición que se ha convertido en una pasión para muchos, por las bellas formas que se logran. Ya lleva siglos de existencia y por ello, tiene una historia muy rica, formando parte esencial de algunas costumbres japonesas. Determinar la fecha del nacimiento del origami (ori, plegado; kami, papel) es algo prácticamente imposible; dadas sus características, debe adoptarse como punto de referencia la elaboración de la primera hoja de papel. Sin embargo, aunque el papiro y el pergamino utilizados con anterioridad al papel eran materiales aptos para la escritura, no lo eran para el arte del plegado, dada la frágil consistencia de los mismos. El papel fue inventado en China alrededor del año 105 de la era cristiana, fruto del desarrollo de los materiales mencionados y por la necesidad de los escribas de perfeccionar su trabajo, reservado en aquellos tiempos a los sacerdotes y a los nobles. Podemos suponer que el plegado del papel haya también nacido en China, alrededor del siglo II, faltando seguros testimonios que precisen el día en que algún escriba se encontró casualmente plegando el papel en el cual estaba trabajando. Plegar el papel resultó, en cuanto se ha dado a suponer, una actitud casual e individual hasta su llegada al Japón, la verdadera patria del origami. Podemos afirmar que toda la cultura japonesa es un continuo desen-volvimiento de armonía y belleza. Pensar en Japón es pensar en forma, en una tierra de modelos, diseños y decoraciones, y en la cual su pueblo se encontró desde siempre inclinado a desentrañar todas las relaciones existentes entre forma y materia. Todas estas características están indisolublemente ligadas al profundo sentido de las tradiciones que sobrevivieron casi intactas, a pesar de que el Japón de hoy sea una poderosa nación industrializada. El ingreso oficial del origami en la cultura japonesa data del siglo IX, cuando aparecieron en los rituales shintoistas los primeros gohei (cintas de papel blanco, plegadas en forma de zigzag) (foto izq.), símbolos de la presencia divina. Por su dificultad de producción y su costo consecuente, el papel por aquel entonces era ya considerado un material noble. También durante las ceremonias nupciales shintoistas era costumbre fijar mariposas de papel en las botellas de sake con las cuales se llenaban las copas utilizadas por los desposados para brindar por la felicidad de su unión. Este par de mariposas (o-chô, o “mariposa macho”, y me-chô, “mariposa hembra”) tenía un profundo significado augural y su plegado estaba reservado solamente a los familiares más cercanos de los nuevos esposos, siendo considerada su confección como una tarea de gran honor. El otro protagonista del primitivo origami fue el noshi, una faja de papel plegado, (foto izq.) utilizada para acompañar los regalos. Para el siglo XII, la práctica del plegado del noshi asumió significados particulares: la elección del papel y su forma de plegarlo en armonía con el obsequio a realizar revelaban el prestigio y la posición social del remitente, mientras que la mayor o menor complejidad de los pliegues demostraba la consideración que el realizador del noshi tenía para con el destinatario del presente. A mediados del Período Edo (1603-1868), el origami pasó de ser una prerrogativa del arte religioso a protagonizar todo tipo de advenimiento mundano. Este pasaje de la utilización sagrada del papel a aquella profana marcó el comienzo de la gran popularidad del origami en el Japón. A partir de dicho período, pequeños objetos de uso doméstico (como agujas o carreteles de hilo) comenzaron a ser colocados en diminutos contenedores de papel (tattô), mientras que las hierbas medicinales eran guardadas en envoltorios (tsutsumi) que realizados en papel permitían mantener el grado justo de humedad natural sin que las hierbas se comprimieran. Asimismo, los niños eran enseñados a confeccionar sus propios juguetes de papel, tales como cascos de guerreros (kabuto) y barriletes a pequeña escala. Desde que comenzó a utilizar el papel, el Occidente debió esperar casi siete siglos para conocer el origami. El encuentro se produjo en 1860, cuando un prestidigitador japonés de gira por Europa maravilló a su público exhibiendo una grulla de papel que movía el cuello al ser tirada de la cola. De esta manera, el origami comenzó a difundirse tímidamente en el mundo, para con el curso del tiempo empezar a ganar más y más adeptos entre los no japoneses. Algunas imagenes del paso a paso: Un consejo: preferentemente los novatos deben usar papel blando , fino y que no sea costoso..es para lograr manejarlo mejor Espero les haya gustado mi aporte!! Saludos!! Atte: yolachiqui T!
El arte del Origami Para los occidentales nos resulta llamativo e ingenioso el arte de crear con papel todo tipo de figuras con determinados dobleces. Es una afición que se ha convertido en una pasión para muchos, por las bellas formas que se logran. Ya lleva siglos de existencia y por ello, tiene una historia muy rica, formando parte esencial de algunas costumbres japonesas. Determinar la fecha del nacimiento del origami (ori, plegado; kami, papel) es algo prácticamente imposible; dadas sus características, debe adoptarse como punto de referencia la elaboración de la primera hoja de papel. Sin embargo, aunque el papiro y el pergamino utilizados con anterioridad al papel eran materiales aptos para la escritura, no lo eran para el arte del plegado, dada la frágil consistencia de los mismos. El papel fue inventado en China alrededor del año 105 de la era cristiana, fruto del desarrollo de los materiales mencionados y por la necesidad de los escribas de perfeccionar su trabajo, reservado en aquellos tiempos a los sacerdotes y a los nobles. Podemos suponer que el plegado del papel haya también nacido en China, alrededor del siglo II, faltando seguros testimonios que precisen el día en que algún escriba se encontró casualmente plegando el papel en el cual estaba trabajando. Plegar el papel resultó, en cuanto se ha dado a suponer, una actitud casual e individual hasta su llegada al Japón, la verdadera patria del origami. Podemos afirmar que toda la cultura japonesa es un continuo desen-volvimiento de armonía y belleza. Pensar en Japón es pensar en forma, en una tierra de modelos, diseños y decoraciones, y en la cual su pueblo se encontró desde siempre inclinado a desentrañar todas las relaciones existentes entre forma y materia. Todas estas características están indisolublemente ligadas al profundo sentido de las tradiciones que sobrevivieron casi intactas, a pesar de que el Japón de hoy sea una poderosa nación industrializada. El ingreso oficial del origami en la cultura japonesa data del siglo IX, cuando aparecieron en los rituales shintoistas los primeros gohei (cintas de papel blanco, plegadas en forma de zigzag) (foto izq.), símbolos de la presencia divina. Por su dificultad de producción y su costo consecuente, el papel por aquel entonces era ya considerado un material noble. También durante las ceremonias nupciales shintoistas era costumbre fijar mariposas de papel en las botellas de sake con las cuales se llenaban las copas utilizadas por los desposados para brindar por la felicidad de su unión. Este par de mariposas (o-chô, o “mariposa macho”, y me-chô, “mariposa hembra”) tenía un profundo significado augural y su plegado estaba reservado solamente a los familiares más cercanos de los nuevos esposos, siendo considerada su confección como una tarea de gran honor. El otro protagonista del primitivo origami fue el noshi, una faja de papel plegado, (foto izq.) utilizada para acompañar los regalos. Para el siglo XII, la práctica del plegado del noshi asumió significados particulares: la elección del papel y su forma de plegarlo en armonía con el obsequio a realizar revelaban el prestigio y la posición social del remitente, mientras que la mayor o menor complejidad de los pliegues demostraba la consideración que el realizador del noshi tenía para con el destinatario del presente. A mediados del Período Edo (1603-1868), el origami pasó de ser una prerrogativa del arte religioso a protagonizar todo tipo de advenimiento mundano. Este pasaje de la utilización sagrada del papel a aquella profana marcó el comienzo de la gran popularidad del origami en el Japón. A partir de dicho período, pequeños objetos de uso doméstico (como agujas o carreteles de hilo) comenzaron a ser colocados en diminutos contenedores de papel (tattô), mientras que las hierbas medicinales eran guardadas en envoltorios (tsutsumi) que realizados en papel permitían mantener el grado justo de humedad natural sin que las hierbas se comprimieran. Asimismo, los niños eran enseñados a confeccionar sus propios juguetes de papel, tales como cascos de guerreros (kabuto) y barriletes a pequeña escala. Desde que comenzó a utilizar el papel, el Occidente debió esperar casi siete siglos para conocer el origami. El encuentro se produjo en 1860, cuando un prestidigitador japonés de gira por Europa maravilló a su público exhibiendo una grulla de papel que movía el cuello al ser tirada de la cola. De esta manera, el origami comenzó a difundirse tímidamente en el mundo, para con el curso del tiempo empezar a ganar más y más adeptos entre los no japoneses. Algunas imagenes del paso a paso: Un consejo: preferentemente los novatos deben usar papel blando , fino y que no sea costoso..es para lograr manejarlo mejor Espero les haya gustado mi aporte!! Saludos!! Atte: yolachiqui T!
El arte del Origami Para los occidentales nos resulta llamativo e ingenioso el arte de crear con papel todo tipo de figuras con determinados dobleces. Es una afición que se ha convertido en una pasión para muchos, por las bellas formas que se logran. Ya lleva siglos de existencia y por ello, tiene una historia muy rica, formando parte esencial de algunas costumbres japonesas. Determinar la fecha del nacimiento del origami (ori, plegado; kami, papel) es algo prácticamente imposible; dadas sus características, debe adoptarse como punto de referencia la elaboración de la primera hoja de papel. Sin embargo, aunque el papiro y el pergamino utilizados con anterioridad al papel eran materiales aptos para la escritura, no lo eran para el arte del plegado, dada la frágil consistencia de los mismos. El papel fue inventado en China alrededor del año 105 de la era cristiana, fruto del desarrollo de los materiales mencionados y por la necesidad de los escribas de perfeccionar su trabajo, reservado en aquellos tiempos a los sacerdotes y a los nobles. Podemos suponer que el plegado del papel haya también nacido en China, alrededor del siglo II, faltando seguros testimonios que precisen el día en que algún escriba se encontró casualmente plegando el papel en el cual estaba trabajando. Plegar el papel resultó, en cuanto se ha dado a suponer, una actitud casual e individual hasta su llegada al Japón, la verdadera patria del origami. Podemos afirmar que toda la cultura japonesa es un continuo desen-volvimiento de armonía y belleza. Pensar en Japón es pensar en forma, en una tierra de modelos, diseños y decoraciones, y en la cual su pueblo se encontró desde siempre inclinado a desentrañar todas las relaciones existentes entre forma y materia. Todas estas características están indisolublemente ligadas al profundo sentido de las tradiciones que sobrevivieron casi intactas, a pesar de que el Japón de hoy sea una poderosa nación industrializada. El ingreso oficial del origami en la cultura japonesa data del siglo IX, cuando aparecieron en los rituales shintoistas los primeros gohei (cintas de papel blanco, plegadas en forma de zigzag) (foto izq.), símbolos de la presencia divina. Por su dificultad de producción y su costo consecuente, el papel por aquel entonces era ya considerado un material noble. También durante las ceremonias nupciales shintoistas era costumbre fijar mariposas de papel en las botellas de sake con las cuales se llenaban las copas utilizadas por los desposados para brindar por la felicidad de su unión. Este par de mariposas (o-chô, o “mariposa macho”, y me-chô, “mariposa hembra”) tenía un profundo significado augural y su plegado estaba reservado solamente a los familiares más cercanos de los nuevos esposos, siendo considerada su confección como una tarea de gran honor. El otro protagonista del primitivo origami fue el noshi, una faja de papel plegado, (foto izq.) utilizada para acompañar los regalos. Para el siglo XII, la práctica del plegado del noshi asumió significados particulares: la elección del papel y su forma de plegarlo en armonía con el obsequio a realizar revelaban el prestigio y la posición social del remitente, mientras que la mayor o menor complejidad de los pliegues demostraba la consideración que el realizador del noshi tenía para con el destinatario del presente. A mediados del Período Edo (1603-1868), el origami pasó de ser una prerrogativa del arte religioso a protagonizar todo tipo de advenimiento mundano. Este pasaje de la utilización sagrada del papel a aquella profana marcó el comienzo de la gran popularidad del origami en el Japón. A partir de dicho período, pequeños objetos de uso doméstico (como agujas o carreteles de hilo) comenzaron a ser colocados en diminutos contenedores de papel (tattô), mientras que las hierbas medicinales eran guardadas en envoltorios (tsutsumi) que realizados en papel permitían mantener el grado justo de humedad natural sin que las hierbas se comprimieran. Asimismo, los niños eran enseñados a confeccionar sus propios juguetes de papel, tales como cascos de guerreros (kabuto) y barriletes a pequeña escala. Desde que comenzó a utilizar el papel, el Occidente debió esperar casi siete siglos para conocer el origami. El encuentro se produjo en 1860, cuando un prestidigitador japonés de gira por Europa maravilló a su público exhibiendo una grulla de papel que movía el cuello al ser tirada de la cola. De esta manera, el origami comenzó a difundirse tímidamente en el mundo, para con el curso del tiempo empezar a ganar más y más adeptos entre los no japoneses. Algunas imagenes del paso a paso: Un consejo: preferentemente los novatos deben usar papel blando , fino y que no sea costoso..es para lograr manejarlo mejor Espero les haya gustado mi aporte!! Saludos!! Atte: yolachiqui T!
Bueno este es un Megapost sobre esta enfermedad llamada "Osteogénesis imperfecta" es una enfermedad que ataca a los huesos, y, a continuacion les voy a explicar todo sobre ella. ¿Qué es la osteogénesis imperfecta? La osteogénesis imperfecta es una enfermedad congénita que se caracteriza porque los huesos de las personas que la sufren se rompen muy fácilmente, con frecuencia tras un traumatismo mínimo e incluso sin causa aparente. Se conocen varios tipos de la enfermedad, y su variación es muy grande de un individuo a otro. Incluso dentro del mismo tipo, puede haber personas con una mayor o menor impregnación. Por decirlo con un ejemplo práctico, algunos pacientes sufren diez fracturas a lo largo de su vida, en tanto que otros pueden llegar a tener varios cientos de ellas. ¿Cuáles son las causas de la osteogénesis imperfecta? La osteogénesis imperfecta se debe a la insuficiente y/o defectuosa formación del colágeno del cuerpo, como consecuencia de un fallo genético. El colágeno es una proteína de los tejidos y su función en la formación de los huesos se puede comparar a la de las nervaduras metálicas en torno a las cuales se monta la estructura de hormigón de una viga. Si la nervadura no es fuerte o no existe, la pieza de hormigón no adquirirá la forma adecuada o será sumamente frágil. La OI no se puede paliar, por tanto, suministrando calcio a los enfermos. No existe hasta el presente ninguna forma de inducir a las células del cuerpo a producir más colágeno o producir colágeno de calidad. ¿Cuál es el pronóstico de la osteogénesis imperfecta? La osteogénesis imperfecta tiene un pronóstico muy variable, dependiendo del grado en que cada individuo esté afectado. La enfermedad en sí no es letal. Sin embargo, las personas afectadas por las formas más graves pueden tener importantes problemas colaterales. Mientras que los afectados por tipos más leves (I y IV según la clasificación de Sillence), en general no tienen más complicaciones que las que impongan sus fracturas y deformaciones óseas, así como las intervenciones quirúrgicas que sean necesarias para tratarlas, la OI del tipo II (siempre según Sillence) suele revestir mucha gravedad y puede llegar a ser letal, debido a las hemorragias que causan las fracturas múltiples en el recién nacido. El tipo III de Sillence tiene un pronóstico variable. En los casos en que la deformación ósea es grave y el volumen torácico es escaso, se pueden presentar problemas de ventilación, que en algunos casos pueden dar lugar a neumonías. En todos los tipos se pueden presentar también problemas cardiovasculares de diferente pronóstico. A pesar de la deformación ósea y la frecuencia de fracturas, la longevidad de una persona afectada por OI es igual a la de cualquier otra. Sus cualidades intelectuales no están mermadas de ninguna forma por la enfermedad y pueden llevar una vida normal, dentro de las limitaciones que imponga el grado de movilidad de cada uno. ¿Cual es la incidencia de la osteogénesis imperfecta? ¿Cómo se transmite? Es difícil determinar el número de personas afectadas por la enfermedad, ya que no hay censos donde consten semejantes datos. Además, podemos partir de la base de que hay gran cantidad de afectados con síntomas muy leves que no han sido ni serán nunca diagnosticados. Yo he leído estimaciones que dicen que la incidencia es de uno entre veinte o treinta mil nacimientos (OIF), y otras según las cuales la cifra debe de andar por un afectado entre cada diez mil nacidos, incluyendo los casos que son diagnosticados a posteriori. En cuanto a la forma en que se transmite, los especialistas no acaban de ponerse de acuerdo. Hasta no hace mucho parecía claro que la forma de transmisión de los tipos I y IV era dominante (estos dos tipos se pueden estudiar en familias donde la enfermedad aparece a lo largo de generaciones). La opinión generalmente aceptada era hasta hace muy poco que tanto el tipo II como el tipo III son el resultado de una transmisión recesiva, pero los más recientes estudios parecen reflejar dudas fundadas sobre esta generalización y apuntan a la posibilidad de que el mosaicismo (error genético en las células germinales de los progenitores) desempeñe un papel importante en la transmisión de los casos más graves de OI. Además, la enfermedad también puede ser el resultado de una mutación espontánea y aparecer en familias sin ningún antecedente. En algunos casos, lo que puede parecer una mutación espontánea no lo es: hay afectados con síntomas tan leves que ignoran su condición hasta que tienen hijos con la enfermedad. En cualquier caso, está claro que los afectados por osteogénesis imperfecta tienen un cincuenta por ciento de probabilidades de transmitirla a su descendencia. Descripción La estructura del hueso es muy parecida al hormigón armado que se utiliza en la construcción de casas. Sobre un marco de barras de acero se vierte el cemento; sin las barras de acero el cemento sería frágil y se rompería al menor movimiento y sin el cemento, las barra no tendrían el apoyo necesario y se doblarían. En los huesos, el papel de las barras de acero lo asume el colágeno y el cemento son los minerales que aporta la sangre (calcio, fósforo, etc.). Los minerales dan fuerza a los huesos y el colágeno proporciona elasticidad. La Osteogénesis Imperfecta (OI) es un trastorno genético que se caracteriza por la fragilidad de los huesos; los huesos pueden fracturarse ante el mínimo golpe o incluso sin causa aparente. El trastorno va a persistir a lo largo de toda la vida de la persona, aunque en muchas de ellas hay un descenso importante del número de fracturas una vez pasada la adolescencia. Debido a las frecuentes fracturas que padecen, en muchas ocasiones se confunde la enfermedad con maltrato infantil. Sintomatología Las características de la enfermedad varían enormemente de un individuo a otro e incluso dentro de los individuos con el mismo tipo de OI. Se han aprobado cuatro formas de enfermedad según características y severidad de la enfermedad: Tipo I Es la forma más frecuente y benigna de la enfermedad. * Los huesos tienen facilidad para fracturarse y la mayor parte de las fracturas se dan antes de la pubertad. * Estatura normal o cercana a la normalidad. * Tono muscular bajo. * La esclerótica (blanco de los ojos) tienen por lo general un tinte azulado. * Cara triangular. * Tendencia a una curvatura anormal de la columna vertebral. * No suele haber deformidad del hueso o ésta es mínima. * En ocasiones los dientes son frágiles. * Puede haber pérdida del oído (más frecuente entre los 20 y 30 años). * La estructura del colágeno es normal pero hay menos cantidad de lo normal. Tipo II Es la forma más severa. Con frecuencia es mortal o la muerte se da poco después del nacimiento normalmente por problemas respiratorios. Hoy en día algunas personas con esta forma de enfermedad han sobrevivido hasta la edad adulta (temprana). * Numerosas fracturas e importante deformidad del hueso. * Baja estatura y los pulmones están poco desarrollados. * El colágeno está mal formado. Los huesos se fracturan con facilidad. A menudo se presentan en el nacimiento e incluso con rayos x se pueden ver las fracturas ya curadas que se dieron antes del nacimiento. * Baja estatura. * La esclerótica tiene un tinte azulado. * Pobre desarrollo muscular de brazos y piernas. * Caja torácica en forma de barril. * Cara triangular. * Curvatura anormal de la columna vertebral. * Posibles problemas respiratorios. *Deformidad de los huesos que a menudo es severa. * Con frecuencia los dientes son muy frágiles. * Posibilidad de pérdida del oído. * El colágeno está formado deficientemente. Tipo IV En cuanto a gravedad se encuentra entre el tipo I y el tipo III. * Los huesos se fracturan con facilidad, sobre todo antes de la pubertad. * Estatura más baja que la media. * La esclerótica suele ser blanca. * Ligera deformidad de los huesos. * Tendencia a una curvatura anormal de la columna vertebral. * Caja torácica en forma de barril. * Cara triangular. * Con frecuencia los dientes son más frágiles. * Pude haber pérdida del oído. * El colágeno se forma de manera incorrecta. Hay que tener en cuenta que la severidad de la fractura no siempre coincide con la intensidad del trauma; puede tener una seria caída y no sufrir ninguna lesión y luego en una actividad cotidiana normal sufrir una fractura. Normalmente, los niños que tienen un crecimiento más rápido tienen mayor número de fracturas; cuando el crecimiento se detiene con la maduración sexual, las células del hueso (osteoblastos), pueden concentrarse en fabricar suficiente matriz del hueso para conseguir una masa ósea suficiente sin necesidad de formar hueso nuevo para el crecimiento, disminuyendo la frecuencia de las fracturas. Aproximadamente el 50% de los pacientes con OI tienen los dientes azulados o marrones. Las anormalidades dentales no pueden ser prevenidas y una mayor limpieza no cambiará la coloración. El primer diente suele aparecer a los 6 meses y si este es normal el resto también lo serán. De todas maneras no suele haber ninguna relación entre decoloración dental y severidad de la enfermedad. El problema es que si los dientes aparecen decolorados suelen gastarse más rápidamente por lo que es importante visitar al dentista con la salida del primer diente y él analizará la necesidad de tratamiento ( ellado para evitar desgaste, etc.). Sentido del oído La pérdida significativa del sentido del oído se da aproximadamente en el 50% de las personas con OI. En el caso de OI tipo I, la forma más frecuente de aparición de pérdida del oído, comienza alrededor del los 20 – 30 años. Hay tres tipos principales del alteración: * Conducción: resultado de un problema físico en el oído medio o externo; puede ser consecuencia de una infección del oído, obstrucción o por fractura de la cadena de huesecillos que se encuentran en el oído, por dentro del yunque. * Neurosensorial: cuando el oído interno no transmite la señal de sonido de forma adecuada al cerebro. * Mezcla: cuando están implicados el oído medio e interno. La pérdida del oído también se puede clasificar según el grado de severidad (leve, moderado, severo y profundo) o por la frecuencia afectada (bajo, alto o todas las frecuencias). Los síntomas que podemos encontrar son: * Dificultad para entender ciertas palabras o partes de palabras. * Preguntas frecuentes al interlocutor para que éste repita las palabras. * Dificultad para entender por teléfono. * Volumen excesivo de la televisión o la radio. * Sensación de entorno ruidoso. Si el mal progresa, la pérdida del sentido del oído puede interferir con la comunicación normal, el rendimiento en el trabajo y en las actividades sociales y personales. Si no se trata adecuadamente puede provocar aislamiento y depresión. Si usted o un miembro de su familia experimenta una pérdida de oído busque ayuda; hay muchos dispositivos de ayuda que pueden serle de gran utilidad; hay organizaciones que pueden ayudarle a encontrar los dispositivos y servicios disponibles. Fracturas Si se sospecha una fractura y no dispone de un traumatólogo de forma inmediata, lo mejor es inmovilizar el miembro y acudir a él cuanto antes. No siempre es posible recibir asistencia médica urgente, por la distancia o cualquier otra circunstancia. En estos casos es útil: Fractura del fémur (hueso del muslo) Hay dos métodos que pueden ser eficaces: * La pierna fracturada se puede proteger (sobre todo para dormir) mediante una toalla pequeña entre las piernas y alrededor de las piernas se coloca una venda elástica. * Cortar un trozo de cartulina y enrollarla al muslo del niño entre la cadera y la rodilla; forrar la cartulina con una tela fina y el conjunto con una venda elástica. Es mejor realizar la operación entre dos personas. Comprobar el posible cambio de color de la piel por si hubiera un problema de circulación de la sangre por tener demasiado apretada la venda. Fractura de húmero (hueso del brazo) Ponga al niño una camisa de manga larga y apoye el brazo firmemente contra el cuerpo limitando el movimiento; póngale otra camisa encima sin introducir el brazo afectado. Fractura de cúbito y radio (huesos del antebrazo) Coloque una revista alrededor del antebrazo (del codo a la muñeca), a continuación una toalla y alrededor una venda elástica. Cuando a su hijo le pongan una escayola hay que tener una serie de precauciones: Cuidados de la escayola o de la inmovilización. El conocimiento de los diferentes métodos que se utilizan para inmovilizar una fractura, así como las técnicas apropiadas para el cuidado del "molde", les permitirá jugar un papel más activo en su recuperación. Los tipos de inmovilización más utilizados son: * Molde de yeso (escayola): muy utilizado por economía y por su flexibilidad. Se usa durante la etapa aguda del proceso de curación de la fractura. * Molde de fibra de vidrio: ligero, fuerte y más resistente al agua que el yeso, si se moja no cambia de forma, pero el acolchado interior permanece mojado e irrita la piel. * Cuando se utiliza la fibra de vidrio junto a un GORE-TEX, la persona puede realizar actividades en las que intervenga el agua, pues no necesita ningún procedimiento de secado. * Entablillado: inmoviliza y mantiene en una determinada posición el hueso o la articulación. Las tablillas se fijan mediante vendas elásticas. * La inmovilización debe permanecer el mínimo tiempo posible. Generalmente se inmoviliza con yeso o con fibra de vidrio; la fibra de vidrio seca más rápidamente, pesa menos y es menos susceptible ante la humedad, pero se amolda peor al miembro. Algunas recomendaciones: * Siga las instrucciones de su médico con respecto a la actividad física. * No la moje. * Procure que no entre suciedad en el interior. * No utilice ningún objeto punzante para aliviar el picor; puede causar heridas en la piel e infecciones. * Frote con alcohol la zona de la piel en contacto con el borde de la escayola (4 veces al día la primera semana). * Eleve la extremidad durante los primeros días, ya que disminuye la inflamación. También puede ser útil aplicar compresas con hielo en la zona afectada. * Inspeccione el molde a diario y póngase en contacto con su médico ante cualquier problema: excesiva presión, frío o entumecimiento, piel en contacto con el molde blanquecina o azulada, olor extraño proveniente del molde, dolor, cosquilleo o entumecimiento. * No intente quitar la inmovilización. Uno de los mayores problemas, en el caso de inmovilización de la pierna, es que la orina moje la escayola; además de la humedad, que nunca es aconsejable, se une el olor, que es dificilísimo de quitar. Puede colocar un pañal especial que se introduce dos o tres dedos en la escayola; otra posibilidad es colocar un plástico alrededor del borde de la escayola en contacto con la zona pubiana. Hay que animar a las personas afectadas para que hagan el mayor ejercicio posible para aumentar la fuerza muscular y la consistencia ósea que puede prevenir las fracturas. La inmovilidad provoca un aumento de la pérdida de hueso y éste es más frágil. La natación es muy aconsejable, ya que el agua permite un movimiento independiente y hay muy poco riesgo de fracturas. Para las personas que puedan, caminar (con ayuda de aparatos o sin ellos) es un ejercicio muy apropiado. Es conveniente consultar con su médico y fisioterapeuta sobre el ejercicio más conveniente para usted. También es beneficioso mantenerse en un peso adecuado, realizando una dieta nutritiva y evitando el alcohol, el tabaco, la cafeína y medicaciones que contengan esteroides, pues pueden aumentar la fragilidad del hueso. Tratamiento de la pérdida de oído Cirugía Las personas con pérdida de oído de tipo conductora, que sea severa y progresiva, pueden recibir ayuda mediante cirugía: estapedectomía: se reemplaza el yunque por una prótesis que permite la transmisión normal de las ondas sonoras al oído interno. Esta operación no debe ser considerada de rutina en los pacientes con OI, debido a la fragilidad del tejido. El empleo del láser para realizar la operación ha mejorado considerablemente el éxito de la operación. Hay muchas cuestiones pre y postoperatorias que hay que evaluar para considerar a una persona como buen candidato para la cirugía. En general, es conveniente que el paciente acuda a un centro especializado con gran experiencia en estapedectomía. Audífonos Aunque los audífonos no curan la pérdida del oído, si pueden proporcionar la amplificación necesaria para tener una audición normal. Medidas Preventivas Debido a la frecuencia de las fractura, la familia necesita de un plan para las situaciones de emergencia que le permita obtener una ayuda inmediata. Consulte con su médico anticipadamente qué medidas debe tomar. Hay que prestar atención a la prevención de la osteoporosis, ya que la gente con OI es más vulnerable a ella que el resto de la gente. Se considera que todos los pacientes tienen una tendencia a desarrollar una baja densidad ósea en algún momento de sus vidas: la osteoporosis es una consecuencia casi universal de la OI. En general, las medidas para prevenir y tratar la osteoporosis en las personas con OI son las mismas que en el resto de la población. Reducir al mínimo la pérdida de hueso La inmovilización en un factor de riesgo crítico para el desarrollo de osteoporosis en las personas. En muchas ocasiones, la inmovilidad es inevitable en las personas con deformidades esqueléticas serias o con fracturas recurrentes. Siempre que sea posible es conveniente realizar actividades como el ejercicio isométrico, levantamiento de pesos, paseos, etc. Eliminar otros factores de riesgo de padecer osteoporosis como pueden ser el tabaco, el consumo excesivo de alcohol y el empleo prolongado de determinadas medicaciones. Las mujeres que se encuentren en la menopausia deben consultar con su médico la conveniencia de administrar suplementos de estrógenos para prevenir la pérdida ósea que se da en este periodo de la vida. Recientemente, varios tipos de bisfosfonatos se han venido utilizando para tratar de aumentar la masa de hueso, con resultados aparentemente buenos, pero todavía es demasiado pronto para sacar conclusiones definitivas. Rehabilitacion. Para poder proporcionar un cuidado óptimo, el médico necesita de ciertos requisitos por parte de sus pacientes y familiares. Es fundamental disponer de una historia médica exhaustiva: fechas de vacunaciones, fracturas, cirugías y otros procedimientos; alergias, complicaciones, reacciones a medicamentos. Así mismo, los padres tienen una serie de derechos que deben pedir a su médico para mantener una relación adecuada: * Información puntual. * Los padres son los que mejor conocen el cuidado diario de su hijo y el médico debe respetarlo. En muchas ocasiones, la preocupación de los padres hace que estén sumamente informados sobre los avances médicos y los médicos deben darles crédito. Además, los niños también deben participar en su propio cuidado y en las discusiones médicas. El médico debe estar dispuesto a participar en esta sociedad formada por él mismo junto con el paciente, los padres y otros profesionales de la salud que serán requeridos puntualmente. Una de las maneras para que el niño pueda alcanzar independencia y confianza es aumentando su movilidad. Existen muchos artículos disponibles para proporcionar mayor movilidad a estos niños, dependiendo de la severidad de la enfermedad, la fuerza muscular del niño; déjese aconsejar por su médico, terapeuta físico y ocupacional. Puede optar, dependiendo de lo comentado, por monopatines especiales en los que el niño apoya el vientre y se propulsa con las manos y rodillas. Otro artículo puede ser el triciclo, también adaptado a sus características, con cinturón de seguridad, tacos de madera en los pedales en caso de corta estatura y apoyo en la espalda para garantizar su estabilidad. Con respecto a las sillas de ruedas, hay muchos modelos disponibles. Desde manuales a eléctricas. Las eléctricas tienen la ventaja de la comodidad y mayor movilidad pero hay una mayor pérdida de masa muscular; analice todas las ventajas e inconvenientes. La inmovilidad debido a la fractura, o simplemente por miedo a una nueva fractura, puede causar una osteoporosis adicional, con aumento de la fragilidad del hueso, y el músculo debilitado, aumentando las posibilidades de nuevas fracturas. Por lo tanto, es recomendable reforzar el músculo cuanto antes; nunca intente limitar el movimiento espontáneo de su hijo, porque cualquier actividad ayudará a reforzar el hueso y el músculo. En cuanto tenga autorización de su médico es importante comenzar con ejercicios de peso y natación, movimientos activos de la cabeza, peso supletorio en los brazos, paseo siempre que sea posible. La natación es un ejercicio sumamente sano y libre de lesiones para los niños con OI. Cuando considere que su hijo ya comienza a ponerse en pie plantéese la posibilidad de ayudarle con andadores. Al principio cualquier mueble fijo le bastará para sujetarse. Los andadores con abrazaderas mantienen al niño en una posición derecha permanente, refuerza los huesos del niño y posiblemente ayude para su crecimiento. El poder dar un paseo es un objetivo para muchos padres y niños; es muy difícil predecir si será posible realizarlo. Muchas personas con OI son capaces de dar un paseo solos, otros se desplazan en silla de ruedas hasta un lugar adecuado para poder dar un corto paseo y en otros casos, después de utilizar andadores, sometidos a intervenciones quirúrgicas y fisioterapia, son incapaces de andar. Siempre hay que proceder como si pudiera andar, hay que tener fe en el niño y permitir que corra los riesgos necesarios para poder alcanzar la máxima movilidad posible. Los terapeutas ocupacionales físicos pueden ayudar a su hijo a desarrollar el adecuado tono muscular, la movilidad y la suficiente fuerza mental para realizar una vida normal y satisfactoria. Fuente: http://salud.discapnet.es/Castellano/Salud/Enfermedades/EnfermedadesDiscapacitantes/O/Osteogenesis%20Imperfecta/Paginas/cover%20osteogenesis.aspx Aca les dejo algunos videos: link: http://www.videos-star.com/watch.php?video=BT20B-ypnHo link: http://www.videos-star.com/watch.php?video=KiqkXBh4fNE link: http://www.videos-star.com/watch.php?video=CAxr1e7K7P0 link: http://www.videos-star.com/watch.php?video=GTpSxlPzC8k link: http://www.videos-star.com/watch.php?video=asKKpeacSIM Si tienen un tiempito pasense por esta pagina se trata sobre un chico llamado Juan que sufre de esta enfermedad, en su pagina cuenta como es su vida, y por todo los momentos que paso. http://yosoyjuani.com.ar/ Aca les dejo algunas imagenes: Bueno, espero que les haya gustado. Saludos! por favore comentenm porqe ya es la 3era vez qe posteo esto y la verdad es qe me costo muchisimo hacerlo..
Ictiosis..Una enfermedad incurable?? Etiología Es una enfermedad que afecta a los niños y se mantiene hasta en la vejez. Se presenta con mayor frecuencia en los meses de invierno, y generalmente se asocia con otras enfermedades cutáneas. Las lesiones son más frecuentes en las extremidades, pero también puede verse afectado el tronco. Síntomas Entre los síntomas se destacan las lesiones cutáneas, producidas por la sequedad, la aparición de "escamas" y una tendencia a engrosarse, y, puede aparecer también un leve picor. Clases de Ictiosis * Ictiosis vulgar: Consisten en escamas finas y blanquecinas, que predominan en el tronco y la cara de extensión de los miembros. La cara, el cuello y las flexuras corporales suelen estar respetadas. Muy a menudo se acompaña de hiperqueratosis folicular en las zonas de extensión de las extremidades, aumento de los pliegues e hiperqueratosis palmo-plantar. Suele mejorar en el verano y empeorar en el invierno. La intensidad de la descamación es muy variable y en los casos leves muchas veces el diagnóstico es consecuencia de un hallazgo en una exploración rutinaria o por otro motivo. La ictiosis vulgar no produce manifestaciones extracutáneas, pero es una asociación frecuente en pacientes con dermatitis atópica. * Ictiosis recesiva ligada al cromosoma X: El inicio suele ser más precoz que en la ictiosis vulgar; en muchos niños las lesiones están presentes desde el nacimiento y en la mayoría son relevantes antes del año de vida. Suele ser más intensa que la ictiosis vulgar, con escamas más gruesas y con mayor frecuencia adquieren una coloración marrón o negruzca. Las zonas más afectadas son también el tronco y las caras de extensión de los miembros, pero también es más habitual la participación de la cara, el cuero cabelludo, el cuello y las flexuras. En cambio, las queratodermia palmoplantar es rara. Al igual que en la ictiosis vulgar, casi todos los casos mejoran durante el verano. Las mujeres embarazadas de fetos con ictiosis ligada a X tienen una incidencia elevada de complicaciones obstétricas y mortalidad perinatal. Se han descrito diversas asociaciones, entre las que se incluyen alteraciones del sistema nervioso central, oculares, genitales y condrodisplasia punctata, pero la gran mayoría de los pacientes no presentan ninguna de ellas y su esperanza de vida es normal. * Ictiosis laminares: Actualmente se admite que es un conjunto de enfermedades con distintas bases bioquímicas y manifestaciones clínicas. El niño puede tener inicialmente el aspecto de un bebé colodión. En las fases evolutivas iniciales suele haber un eritema como base del cuadro ictiosiforme , que en algunos casos tiende a atenuarse o se hace imperceptible con el tiempo. La descamación es generalizada, con tamaño de escamas muy variable. El ectropión, el eclabión, la queratodermia palmoplantar y la alopecia son hallazgos frecuentes. Las principales asociaciones descritas son talla baja y retraso mental. * Eritrodermia ictiosiforme congénita ampollosa: Aunque con menos frecuencia que las ictiosis laminares, puede presentarse como un bebé colodión. La mayoría de las veces el neonato está eritrodérmico y se observan áreas erosivas y denudadas; la observación de ampollas íntegras es posible, pero rara. La eritrodermia tiende a persistir, aunque se puede atenuar y las erosiones y las ampollas van disminuyendo con el tiempo, a la vez que aumenta la hiperqueratosis, sobre todo en las zonas flexurales, donde puede hacerse verrugosa. En conjunto hay una tendencia a la mejoría con la edad. * Bebé colodión: Es una forma de presentación de diversos tipos de ictiosis, sobre todo de las ictiosis laminares. Algunos casos pueden evolucionar hacia la curación. Los niños nacen con una piel eritematosa y con el aspecto de estar envueltos en celofán. Esta envoltura superficial tiende a agrietarse y más tarde a desprenderse en grandes láminas, tras lo cual se instauran las características clínicas del proceso de base. Es habitual que exista ectropión y eclabión y puede causar dificultad respiratoria por constricción torácica y abdominal. * La Ictiosis Arlequín: es una enfermedad de la piel extremadamente rara del grupo de las llamadas genodermatosis (grupo de dermatosis hereditarias con trastornos metabólicos). Es la forma de ictiosis congénita más grave, se hace evidente ya desde el nacimiento y debe su nombre al aspecto que tienen los recién nacidos con la enfermedad, que recuerda a un disfraz de arlequín. Tratamiento El tratamiento habitual de la Ictiosis incluye las cremas tópicas. Aquí, la variedad y eficacia de cada una de ellas varía en cada paciente. Cada uno debe encontrar las que mejor se adaptan a su tipo y grado de Ictiosis. Estas cremas suelen ser de tres tipos: * Humectantes: cremas y lociones que penetran y humedecen la piel. * Queratolíticos: promueven la exfoliación de la piel. * Emolientes: son lubricantes que humedecen la piel y sellan la humedad en el interior. En los casos de Ictiosis grave, como la Ictiosis Congénita grave o la Enfermedad de Darier está indicado el tratamiento con Acitretina, producto sintético relacionado estructuralmente con la vitamina A, cuya respuesta terapéutica normal consiste en la descamación, seguida de reepitelización más normal. El uso de este medicamento siempre e inexcusablemente, deberá estar controlado por el especialista. Diagnóstico El diagnóstico se confirma mediante biopsia y examen al microscopio electrónico, para poder diferenciarla de la dermatitis atópica, afección muy común y banal, así como de otros tipos de ictiosis. Herencia Las ictiosis puede ser hereditaria y adquirida: Las ictiosis hereditarias son raras y por lo general aparecen durante la infancia y se mantienen de por vida, siendo frecuentemente formas de afectación leve y que tienden a mejorar durante los meses de verano, aunque también pueden existir algunas formas clínicas muy severas. Las ictiosis hereditarias se caracterizan por el gran acumulo de escamas sobre la superficie cutánea y se clasifican en función de criterios gen éticos y clínicos. Las ictiosis adquiridas aparecen asociadas a otras enfermedades, sobre todo renales y pueden comenzar en cualquier época de la vida, pueden constituir una manifestación precoz de algunas enfermedades sistémicas (lepra, hipotiroidismo, linfoma, SIDA). La descamación seca puede ser fina y localizarse exclusivamente en el tronco y las piernas, o ser gruesa y difusa. La biopsia (operación que consiste en extirpar en el individuo vivo un fragmento de órgano o de tumor con objeto de someterlo a examen microscópico) de la piel ictiósica no suele resultar diagnóstica, aunque existen excepciones, como la sarcoidosis, en la que se producen escamas gruesas en las piernas y presencia de granulomas típicos en la biopsia. La ictiosis vulgar es el tipo más frecuente de ictiosis y la padecen el 30-40% de pacientes con dermatitis atópica. Discusión de bebe colodion: La patogénesis es desconocida. En dos casos de bebé colodión que curaron sin secuelas, Raghunath y cols encontraron una mutación en la transglutaminasa-13. La mortalidad y morbilidad esta aumentada. La mortalidad ha descendido desde el 50% en los años 60 hasta el 11% en los años 80. La causa de muerte más frecuente es la sepsis tras una infección cutánea4 . La disminución de la mortalidad se debe tanto al avance de la neonatología como al mejor conocimiento y prevención de las complicaciones como ser la deshidratación por perdida transepidérmica de agua, las alteraciones electrolíticas, (hipernatremia, hipercalcemia) y alteraciones en la termorregulación (hipo e hipertermia)5 . El pronóstico del bebe colodión es imposible de determinar a nivel individual, ya que puede evolucionar hacia la curación espontánea (4 al 24% de los casos) o bien hacia algún tipo de ictiosis, más frecuentemente hacia la ictiosis lamelar en un 50%, eritrodermia congénita no ampollosa (11%) o bien hacia formas más raras como; tricotiodistrofia o síndrome de Tay, síndrome de Sjögren-Larsson, enfermedad de Conradi, hipotiroidismo congénito6 , o enfermedad de Gaucher. En lo referente al tratamiento: El paciente debe permanecer en incubadora, con un alto grado de humedad y a temperatura constante sin variaciones bruscas, vigilancia del grado de hidratación y prevenir las infecciones. El tratamiento tópico es importante, pero se deben evitar las sobre infecciones producidas por estreptococos y estafilococos. Es importante evitar los queratolíticos como el ácido salicílico o la urea, pues al estar aumentada la absorción percutánea se puede producir intoxicación por ácido salicílico, e hiperuricemia, algunos autores proponen el tratamiento tópico con Vitamina A ácida y oclusión con plástico por 24 a 48 horas. Debe instituirse tempranamente lubricantes oculares tópicos, para evitar el riesgo de ulceras cornéales, producidas por el ectropión resultante de la piel tensa que evierte los bordes palpebrales.7 Les dejo algunas imagenes: En este link van a poder ver un pqueño articulo de diario sobre un chico que padecio de Ictiosis y cumplio su sueño, ser Carabinero. http://www.diarioviregion.cl/modules.php?name=News&file=print&sid=5115 Fuente: http://salud.discapnet.es/Castellano/Salud/Enfermedades/EnfermedadesDiscapacitantes/I/Ictiosis/Paginas/cover%20ictiosis.aspx?tema=1&rd=04062008170553 Saludos atte: Camila (Yolachiqui) porfavor comenten es la 3' vez qe lo posteo y en todas recibi 1 solo comentario.
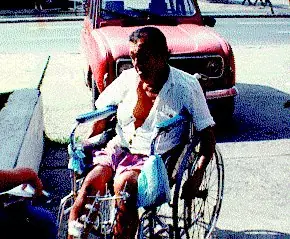
¿Cuales son las principales discapacidades? Existen cinco principales tipos de discapacidad, clasificadas según los ámbitos del ser humano que afectan: * Discapacidad auditiva... * Discapacidad física.... * Discapacidad mental... * Discapacidad psíquica * Discapacidad visual Conceptos * Deficiencia: Es toda pérdida o anormalidad de una estructura o función sicológica, fisiológica o anatómica. * Discapacidad: Es toda restricción o ausencia (debido a una deficiencia) de la capacidad de realizar una actividad en la forma o dentro del margen que se considera normal para un ser humano. * Minusvalía: Es una situación desventajosa para un individuo determinado, consecuencia de una deficiencia o de una discapacidad, que limita o impide el desempeño de un papel que es normal en su caso (en función de su edad, sexo y factores sociales y culturales). Discapacidades Alzheimer Discapacidad Psíquica Lesiones Medulares Ataxia Esclerosis Lateral Amiotrófica (ELA) Osteogénesis Imperfecta Cardiopatías Esclerosis Múltiple Parálisis Cerebral Distonía Extrofia Vesical Parkinson Discapacidad Auditiva Fibrosis Quística Poliomielitis Discapacidad Mental Insuficiencia Renal Crónica Síndrome de Down Discapacidad Visual Ø Alzheimer ¿Qué es el mal de Alzheimer? Científicamente, se define como una demencia progresiva y degenerativa del cerebro. Provoca en un principio un deterioro en la memoria inmediata. Las neuronas que controlan la memoria y el pensamiento se van deteriorando, interrumpiendo el paso de mensajes entre ellas. Las células desarrollan cambios distintivos: placas seniles y neurofibrilares (degeneraciones del tejido cerebral). La corteza cerebral (principal origen de las funciones intelectuales) se atrofia y encoge. ¿Cómo se diagnostica? En la actualidad, no existe una sola prueba diagnóstica para la Enfermedad de Alzheimer. Cuando se observen los síntomas, se debe obtener una evaluación física, psiquiátrica y neurológica completa, realizada por un médico con experiencia en el diagnostico de trastornos de demencia. ¿Hay tratamiento? Esta enfermedad no tiene cura, pero tiene tratamientos que se utilizan para paliar la situación. Hay medicamentos que ayudan a desacelerar el deterioro de la acetilcolína, un producto químico necesario para la comunicación de cerebro-célula, este se agota gravemente en Alzheimer. También para el síntoma de deambulación, agitación y la alteración del sueño. El sentirse cuidado con amor, con muchísima paciencia y compresión, es la mejor medicación que podemos suministrarles. Ø Ataxia ¿Qué es la Ataxia? Es una enfermedad degenerativa de origen neurológico. Gran parte de las Ataxias son de transmisión hereditaria, ya sea de forma dominante o recesiva. Este es el motivo por el que a los afectados se les plantean serias dudas a la hora de enfrentarse a una futura paternidad. ¿Cómo se diagnostica? Los primeros síntomas frecuentemente son tropiezos o un caminar similar al de alguien ligeramente ebrio, seguido por inestabilidad e incoordinación de movimientos en los brazos y las manos. En algunos pacientes los síntomas progresan con bastante rapidez durante un periodo de 10 a 15 años, o despacio en otros pacientes durante un espacio de 20 años o más. Los afectados en el futuro necesitarán una silla de ruedas que les proporcionará independencia de movimientos. La Ataxia es una vida reducida por la enfermedad. Los afectados experimentarán dificultades para permanecer de pie, caminar, correr y mantener el equilibrio. ¿Hay tratamiento? Esta enfermedad no tiene cura. Ø Cardiopatías Comunicación interauricular. Las comunicaciones interauriculares (CIA) se producen por defectos en el tabique interauricular, lo cual hace que la sangre fluya desde la aurícula izquierda hacia la aurícula derecha. La localización del defecto en el tabique interaricular es variable, por lo que se suelen distinguir varios tipos. El defecto del tipo seno venoso se localiza en la parte alta del tabique interauricular, próximo a la entrada de la cava superior. La CIA tipo ostium secundum es la más frecuente y se localiza en la parte media del tabique interauricular. La CIA tipo ostium primum se localiza en la parte baja del tabique interauricular, inmediatamente adyacente a las válvulas aurículo-ventriculares, y es una forma de defecto del canal aurículo-ventricular. Un caso especial de cortocircuito izquierda-derecha a nivel auricular es el que se produce por la ausencia congénita del tabique interauricular, lo que da lugar a una aurícula única, que es una anomalía poco frecuente. Etiología. La causa de los defectos del tabique interaricular no es bien conocida. Lo más frecuente es que se encuentren de forma aislada, sin causa evidente ni enfermedad concomitante. En algunos casos, sin embargo, se encuentra una etiología. Así, la rubeola de la madre durante el embarazo se ha relacionado con defectos del tabique interaricular en el hijo. Diversos síndromes genéticos, como el de Ellis-van Cleveld y el de Holt-Oram muestran una incidencia aumentada de CIA, al igual que en el síndrome TAR, que cursa con trombocitopenia y agenesia o hipoplasia del radio. La aurícula única suele observarse en pacientes con síndrome de Ellis-van Cleveld o asociada a asplenia o polisplenia. En la trisomía 21 o síndrome de Down se observa una incrementada incidencia de defectos del canal aurículo-ventricular. Además, existe una forma familiar de defectos del canal aurículo-ventricular, que se transmite mediante herencia autosómica dominante. Por otro lado, la CIA tipo ostium secundum presenta 2 formas familiares, que se transmiten con una herencia autosómica dominante y que no se asocian a anomalías extracardiacas. La forma familiar más frecuente cursa con CIA tipo ostium secundum y bloqueo aurículo-ventricular. La otra forma familiar es poco frecuente y sólo muestra el defecto del tabique interaricular sin ninguna otra anomalía acompañante. Ø Discapacidad Auditiva Es la pérdida total de la audición. La prevención de la sordera es relativamente difícil debido a las numerosas causas que la provocan en los distintos períodos: prenatal, perinatal y posnatal. La causa hereditaria o genética es la más importante y desgraciadamente poco previsible. En el período perinatal, la sordera se produce por problemas de partos anormales por causa fetal o materna. Dentro de las causas posnatales de sorderas, ocupa un lugar muy importante aún la meningitis bacteriana. En segundo lugar, las otitis medias producen habitualmente un deterioro paulatino de la audición. En tercer lugar, la sordera es producida por ruidos de alta intensidad. Según el Censo de 1992, en Chile hay 59.048 personas con sordera total. La audiometría permite precisar el grado de sordera (evaluado en decibelios de pérdida), y su tipo. Así se distingue las sorderas de transmisión, que afectan al conducto auditivo externo (oído externo), el tímpano, la caja del tímpano y la cadena de huesos (oído medio), y las sorderas de percepción, que corresponden a una lesión de la cóclea o del nervio auditivo (oído interno). Ø Discapacidad Mental Según la American Association of Mental Retardation, el retraso mental alude a limitaciones sustanciales en el funcionamiento actual. Se caracteriza por un funcionamiento intelectual significativamente inferior a la media, que generalmente coexiste junto a limitaciones en dos o más de las siguientes áreas de habilidades de adaptación: comunicación, auto-cuidado, vida en el hogar, habilidades sociales, utilización de la comunidad, auto-dirección, salud y seguridad, habilidades académicas funcionales, tiempo libre y trabajo. A menudo, junto a limitaciones especificas coexisten potencialidades en otras áreas adaptadas o capacidades personales. Algunos ejemplos de deficiencias que pueden derivar en una discapacidad mental son el síndrome de Down, el síndrome de X Frágil, el síndrome de Wesn y la Fenilcetonuria. Sin embargo, existen muchas más. Ø Discapacidad Psíquica Se considera que una persona tiene discapacidad psíquica cuando presenta "trastornos en el comportamiento adaptado, previsiblemente permanentes". ¿Cuáles son las causas? La discapacidad psíquica puede ser provocada por diversos trastornos mentales, como la depresión mayor, la esquizofrenia, el trastorno bipolar; los trastornos de pánico, el trastorno esquizomorfo y el síndrome orgánico. Ø Discapacidad Visual ¿Qué es la Ceguera? Según la Organización Mundial de la Salud (OMS), es aquella visión menor de 20/400 ó 0.05, considerando siempre el mejor ojo y con la mejor corrección. Se considera que existe ceguera legal cuando la visión es menor de 20/200 ó 0.1 en el mejor ojo y con la mejor Corrección. ¿Qué es baja Visión? Es una visión insuficiente, aun con los mejores lentes Correctivos, para realizar una tarea deseada. Desde el punto de vista funcional, pueden considerarse como personas con baja visión a aquellas que poseen un resto visual suficiente para ver la luz, orientarse por ella y emplearla con propósitos funcionales. ¿Cuáles son las Causas? Aunque la ceguera puede ser provocada por algún accidente, también existen numerosas enfermedades que la desencadenan: Catarata, glaucoma, leucomas cornéales, retinopatía diabética, retinopatía del prematuro, catarata y glaucoma congénitas, atrofia óptica, distrofia retinal y retinosis pigmentaria, entre otras. ¿Se puede prevenir? Existen diversas maneras de prevenir la discapacidad visual, como el evitar accidentes del tránsito, del trabajo y enfermedades ocupacionales; atención adecuada del embarazo; detección y registro de deficiencias en los recién nacidos y el asesoramiento genético a las familias en los casos de enfermedades hereditarias. La consulta oftalmológica precoz cuando hay antecedentes en la familia también contribuye a la prevención. Ø Distonía Muscular ¿Qué es Distonía? Distonía es tanto el síntoma como el nombre de un grupo de enfermedades, precisamente denominadas distonías. El síntoma o manifestación física, consiste en permanentes contracciones involuntarias de los músculos de una o más partes del cuerpo. Esto a menudo se manifiesta en torsiones o deformaciones de esa parte del cuerpo. El trastorno es secundario a la disfunción del sistema nervioso central, probablemente en aquellas partes del cerebro llamados ganglios basales. En la de tipo primaria o distonía sin complicaciones, no hay alteración de la conciencia, de las sensaciones o de la función intelectual. La distonía puede estar acompañada de temblor, algunas veces parecido a los temblores que generalmente se observan en personas de edad, o por un temblor de tipo severo, de calidad irregular e inestable. ¿Qué hacer si se tiene un diagnóstico de Distonía? Si se considera un diagnóstico de distonía, lo primero que se debe hacer es revisar cuidadosamente el árbol genealógico de la familia, buscando personas que tengan distonía focal o generalizada. Aún el más simple "cuello torcido" (tortícolis) debe ser tomado en cuenta. Como también cualquier tipo de temblor. Esta información es muy significativa para un examen genético. En segundo lugar, se debe estudiar el tipo de terapia médica a seguir. Aquí un neurólogo con una considerable experiencia debe ser su guía. Muchos medicamentos están disponibles. Cualquier medicamento puede ser moderadamente efectivo, o medianamente efectivo, o simplemente no ser de ninguna ayuda. Sin embargo, la persistencia en probar medicamentos puede ser finalmente recompensada. Terapia con medicamentos Algunos de los medicamentos que su doctor puede considerar incluyen: Artane (trihexyphenidyl), Cogentin (benztropine), Valium (diazepam), Klonapin (clonazepam), Lioresal (baclofen), Tegretol (carbamazepine), Sinemet o Madopar (levodopa), Parlodel (bromocriptine), Symmetrel (amantadine). Ø Esclerosis Lateral Amiotrófica (ELA) ¿Qué es la ELA? La ELA es una enfermedad neuromuscular en la que las células nerviosas, las motoneuronas que controlan el movimiento de la musculatura voluntaria, gradualmente disminuyen su funcionamiento y mueren, provocando debilidad y atrofia muscular. Estas motoneuronas se localizan en el cerebro y en la médula espinal. ¿Cuál es la causa de la ELA? La causa de la ELA es desconocida. En cualquier caso, es un hecho que el conocimiento que se tiene del funcionamiento del sistema nervioso es cada vez mayor, y aumenta cada año, gracias a la utilización de herramientas más sofisticadas en el ámbito de la biología molecular, ingeniería genética y bioquímica. Todo esto nos hace albergar mayores esperanzas a la hora de pensar en un pronto descubrimiento de su origen. ¿Cómo se diagnostica la ELA? El diagnóstico es fundamentalmente clínico, es decir, no existe ninguna prueba específica que dé el diagnóstico definitivo. Después de que se haya confirmado el diagnóstico de ELA, se deben practicar numerosas pruebas de distinto tipo para descartar otras enfermedades que pueden simular la ELA. Con estos test, el estudio de la historia clínica del paciente y un detenido examen neurológico, los especialistas suelen llegar al diagnóstico definitivo. De entre las pruebas que se deben realizar para su diagnóstico, destacan una resonancia nuclear magnética, cerebral o espinal, un estudio electromiográfico de la función neuromuscular y una batería de análisis de sangre y de orina específicos. Nosotros recomendamos siempre que los pacientes tengan un segundo diagnóstico realizado por un médico con experiencia en ELA, con el fin de reducir el número de diagnósticos incorrectos. En muchas ocasiones, el diagnóstico definitivo puede tardar varios meses en producirse, aún después de realizar todos los test pertinentes y observar atentamente la evolución de los síntomas. ¿Existe algún tratamiento? Por ahora no existe ningún tratamiento probado contra la ELA Sin embargo, el reciente descubrimiento de determinados factores de crecimiento neuronal y de agentes bloqueantes del glutamato, se han mostrado prometedores en la detención de la progresión de la enfermedad, aunque no existe aún ningún fármaco que la cure. Sí existen fármacos para combatir el cortejo sintomático que acompaña a la enfermedad, como son los calambres, la espasticidad, las alteraciones en el sueño o los problemas de salivación. Existen numerosas estrategias muy eficaces para cuando aparecen las alteraciones respiratorias o cuando surgen problemas relacionados con las secreciones. Los fisioterapeutas, terapeutas ocupacionales y logopedas, son los profesionales encargados de asegurar la independencia funcional a través del ejercicio y la utilización de los equipos técnicos oportunos. Ø Esclerosis Múltiple ¿Qué es la esclerosis múltiple? La esclerosis múltiple (EM) es una enfermedad del sistema nervioso central (SNC) en la que se diferencian dos partes principales: cerebro y médula espinal. Envolviendo y protegiendo las fibras nerviosas del SNC hay un material compuesto por proteínas y grasas llamado mielina que facilita la conducción de los impulsos eléctricos entre las fibras nerviosas. En la EM la mielina se pierde en múltiples áreas dejando en ocasiones, cicatrices (esclerosis). Estas áreas lesionadas se conocen también con el nombre de placas de desmielinización. La mielina no solamente protege las fibras nerviosas sino que también facilita su función. Si la mielina se destruye o se lesiona, la habilidad de los nervios para conducir impulsos eléctricos desde y al cerebro se interrumpe y este hecho produce la aparición de síntomas. Afortunadamente la lesión de la mielina es reversible en muchas ocasiones. La EM no es ni contagiosa, ni hereditaria, ni mortal. ¿Cuáles son los síntomas de la EM? Los síntomas incluyen debilidad, hormigueo, poca coordinación, fatiga, problemas de equilibrio, alteraciones visuales, temblor, espasticidad o rigidez muscular, trastornos del habla, problemas intestinales o urinarios, andar inestable (ataxia), problemas en la función sexual, sensibilidad al calor, problemas de memoria a corto plazo y ocasionalmente problemas de juicio o razonamiento (problemas cognoscitivos). Hay que remarcar que la mayoría de personas con EM no tienen todos estos síntomas. Ø Espina Bífida Malformación sin causa Es una malformación congénita que origina diversas alteraciones en el organismo y que tiene distintos grados de afección. Espina Bífida (EB) es una de las malformaciones más graves del tubo neural, compatibles con una vida prolongada. Se da en el nacimiento por una falta de cierre o fusión de varios arcos vertebrales. El defecto se origina precozmente en el primer mes de gestación. Las causas son desconocidas, pero se cree que es la resultante de una combinación de factores genéticos y ambientales. Esta anomalía puede estar localizada en cualquier zona de la columna vertebral y dependiendo de su grado de gravedad hay tres tipos diferentes de EB: espina Bífida oculta se halla en la parte inferior de la columna y consiste en una pequeña obertura de un arco vertebral. Ø Extrofia Vesical ¿Qué es la Extrofia Vesical? La palabra extrofia se deriva de la palabra griega ekstriphein que literalmente significa vuelta o giro de dentro a fuera. La extrofia de la vejiga es una malformación de la vejiga y uretra, en la que la vejiga ha girado de adentro a fuera. La uretra, que es el canal que lleva la orina fuera del cuerpo, no se ha formado completamente. Esto se llama epispadias. En chicos, el pene tiene apariencia aplastada y podría estar levantado (alzado) hacia el abdomen. En las niñas, la uretra abierta es localizada entre un clítoris dividido y el labio menor. ¿Qué determina la magnitud de la Extrofia? La magnitud de la extrofia depende de cuan pequeña o grande es la apertura. Uno puede pensar en la extrofia como un espectro de condiciones que van desde la forma más apacible, epispadias (sólo un defecto en la uretra o apertura), a la forma más severa, extrofia cloacal (defecto en la uretra, vejiga e intestino), estando la extrofia vesical clásica (defecto en la uretra y la vejiga) en el medio de las dos anteriores. ¿Qué causa la Extrofía Vesical? La ciencia médica no puede explicar el por qué un embrión puede desarrollar extrofia vesical. No se debe a nada que el padre o la madre hizo o no hizo durante el embarazo. Por otro lado, no es simplemente una condición heredada. Ø Fibrosis Quística ¿Qué es la Fibrosis Quística? La Fibrosis Quística es una enfermedad crónica, en la mayoría de los casos degenerativa y de carácter genético (no se adquiere a lo largo de la vida) y hereditaria, trasmitida conjuntamente por ambos progenitores y que ocasiona una patología de tipo evolutivo que varía de una persona a otra. Debe negarse rotundamente que se trate de una enfermedad contagiosa, por lo que el contacto con una personas afectada no reviste peligro de contagio. Ø Insuficiencia Renal Crónica ¿Qué es la Insuficiencia Renal Crónica? Se entiende por insuficiencia renal la pérdida de la función de los dos riñones. Habitualmente, esta pérdida se produce a la vez en ambos y es importante destacar que un sólo riñón sin problemas es suficiente para mantener una función completamente normal. La insuficiencia renal puede ser aguda cuando aparece de forma brusca y normalmente tiende a recuperarse, y crónica cuando el fallo de función de los riñones se produce de forma lenta y progresiva, sin posibilidades de recuperación. Ø Lesiones Medulares Cualquier daño a la médula espinal es una lesión muy compleja. Cada lesión es diferente y puede afectar el cuerpo en varias formas diferentes. Este en un breve resumen de los cambios que suceden después de una lesión en la médula espinal. Esto dice como la médula espinal funciona y qué puede pasar con el cuerpo después de una lesión en la médula espinal. La Médula Espinal Normal Los nervios son estructuras similares a un cordón compuestos de muchas fibras nerviosas. La médula espinal tiene muchas fibras nerviosas espinales. Las fibras nerviosas transportan mensajes entre el cerebro y las diferentes partes del cuerpo. Los mensajes pueden ser relacionados con el movimiento, diciéndole a alguna parte del cuerpo que se mueva. Otras fibras nerviosas llevan mensajes de sensación o tacto desde el cuerpo hacia el cerebro, como el calor, el frío o el dolor. El cuerpo también tiene un sistema nervioso autonómico. El controla las actividades involuntarias del cuerpo como la presión sanguínea, la temperatura corporal y el sudor. Estas fibras nerviosas constituyen el sistema de comunicación del cuerpo. La médula espinal puede ser comparada a un cable telefónico. Conecta la oficina principal (el cerebro), con otras oficinas particulares (las partes del cuerpo) por medio de líneas telefónicas (las fibras nerviosas). La médula espinal es el camino que los mensajes usan para viajar entre el cerebro y las diferentes partes del cuerpo. Debido a que la médula espinal es una parte vital de nuestro sistema nervioso, está rodeado y protegido por huesos llamados vértebras. Las vértebras, o huesos de la espalda, están colocadas una arriba de la otra y se llama la columna vertebral o la columna espinal. La columna vertebral es el soporte número uno del cuerpo. La médula espinal realmente pasa por en medio de las vértebras. La médula espinal tiene aproximadamente 45 cm. de largo. Se extiende desde la base del cerebro, continúa hacia abajo de la mitad de la espalda, aproximadamente hasta la cintura. El haz de fibras nerviosas que constituyen la médula espinal por si mismo son las neuronas motoras superiores (UMN). Los nervios espinales se ramifican desde la médula espinal hacia arriba y hacia abajo de el cuello y la espalda. Estos nervios, neuronas motoras inferiores (LMN), salen entre cada vértebra y alcanzan todas las partes del cuerpo. La médula espinal termina cerca de la línea de la cintura. Desde este punto, las fibras nerviosas espinales bajas continúan hacia abajo a través del canal espinal hasta el sacro o coxis. La columna espinal está dividida en cuatro secciones o partes. La porción superior, es nombrada el área cervical, tiene siete vértebras cervicales. La sección que sigue, la dorsal, incluye el área del pecho y tiene doce vértebras dorsales. La sección baja de la espalda es nombrada el área lumbar. Hay cinco vértebras lumbares. La sección final tiene cinco vértebras sacras y es nombrada el área sacra. Los huesos en la sección sacra, en realidad están fusionados en un solo hueso. Ø Osteogénesis Imperfecta ¿Qué es la Osteogénesis Imperfecta? La osteogénesis imperfecta es una enfermedad congénita que se caracteriza porque los huesos de las personas que la sufren se rompen muy fácilmente, con frecuencia tras un traumatismo mínimo e incluso sin causa aparente. Se conocen varios tipos de la enfermedad, y su variación es muy grande de un individuo a otro. Incluso dentro del mismo tipo, puede haber personas con una mayor o menor impregnación. Por decirlo con un ejemplo práctico, algunos pacientes sufren diez fracturas a lo largo de su vida, en tanto que otros pueden llegar a tener varios cientos de ellas. ¿Cuál es la causa de la Osteogénesis Imperfecta? La osteogénesis imperfecta se debe a la insuficiente y/o defectuosa formación del colágeno del cuerpo, como consecuencia de un fallo genético. El colágeno es una proteína de los tejidos y su función en la formación de los huesos se puede comparar a la de las nervaduras metálicas en torno a las cuales se monta la estructura de hormigón de una viga. Si la nervadura no es fuerte o no existe, la pieza de hormigón no adquirirá la forma adecuada o será sumamente frágil. Ø Parálisis Cerebral ¿Qué es la Parálisis Cerebral? Es un conjunto de desórdenes cerebrales que afecta el movimiento y la coordinación muscular. Es causada por daño a una o más áreas específicas del cerebro, generalmente durante el desarrollo fetal, pero también puede producirse justo antes, durante o poco después del nacimiento, así como en la infancia. Existen diversos grados de parálisis cerebral. Además, tradicionalmente se distinguen cuatro tipos de Parálisis Cerebral: Espástica (la manifestación más común, en que los músculos permanecen tensos, parecen rígidos y los movimientos voluntarios como el caminar son difíciles; a veces pueden); Disquinética (que se caracteriza por movimiento involuntario de la cara, las manos y otras partes del cuerpo); Atáxica (que causa problemas de equilibrio y coordinación, particularmente al caminar) y Mixta (que puede combinar cualquiera de los tipos anteriores). ¿Cuáles son las causas? Puede ser producida por una serie de factores. Una causa importante es la falta de oxígeno en el cerebro durante la etapa fetal o de recién nacido, que puede deberse a una separación prematura de la placenta desde la pared del útero, mala posición natal del bebé, un parto muy largo o abrupto o interferencia de la circulación en el cordón umbilical. También puede causar Parálisis Cerebral una incompatibilidad sanguínea entre la madre y el infante o la infección de la madre con virus y otros microorganismos que atacan el sistema nervioso del infante. Una herida en el cráneo o falta de oxigenación por inmersión también son causas frecuentes, como resultado de accidentes. ¿Cuáles son las características? Dependiendo de qué áreas del cerebro tiene dañadas; una persona con Parálisis Cerebral puede presentar algunas de las siguientes características: Tensión muscular o espasmos; movimientos involuntarios; sensación y percepción anormales; limitación visual, auditiva o del habla; tercianas; retraso mental. Quienes tienen Parálisis Cerebral también pueden presentar dificultades para comer para controlar la vejiga y los esfínteres, para respirar debido a problemas posturales, escaras, o discapacidades para el aprendizaje. Ø Parkinson El hallazgo de un factor tóxico-ambiental como responsable de alteraciones parkinsonianas, ha producido un importante impulso en el ámbito de la investigación de la causa de esta enfermedad. ¿Cuáles son los síntomas? La enfermedad de Parkinson se manifiesta básicamente por la presencia de temblor, rigidez de los músculos y lentitud y dificultad para iniciar el movimiento. Estos síntomas pueden aparecer aislados o combinados, pudiendo predominar en una parte del cuerpo o bien ser más marcado un síntoma sobre los demás, de forma que hay gran variación de un enfermo a otro. En los comienzos pueden aparecer molestias muy variables, difíciles de relacionar con la enfermedad. No es raro que los pacientes acudan inicialmente al traumatólogo, aquejando dolores en las articulaciones, o al psiquiatra buscando el tratamiento de un estado depresivo. Ø Poliomietis El virus usualmente entra al cuerpo por el conducto alimentario y afecta a varias partes del sistema nervioso central. Los períodos de la incubación tienen una duración de aproximadamente entre 4 a 35 días. Síntomas tempranos incluyen fatiga, dolor de cabeza, fiebre, vómitos, estreñimiento, tiesura del cuello, ó, menos normalmente, diarrea y dolor en las extremidades. Como las células no se reemplazan, los nervios que controlan el movimiento muscular se destruyen, y la infección del poliovirus puede causar parálisis permanente. Cuando las células atacadas son en centros respiratorios, el control de la respiración, se destruye y se debe aislar al enfermo en un pulmón férrico (Respiración Artificial). Aspectos clínicos El cuadro más característico se compone de fiebre elevada de instauración brusca, dolores musculares y parálisis bastante particulares. La aparición de las parálisis es repentina y su topografía anárquica. El déficit motor afecta a cualquier músculo, de manera asimétrica y desigual. A él se asocia una disminución de los reflejos osteotendinosos y una rápida tendencia a la atrofia muscular. Un detalle importante es que en esta enfermedad no existen nunca trastornos de la sensibilidad. Ø Síndrome de Down El Síndrome de Down (SD) es un accidente genético al que cualquier persona esta expuesta. Es una alteración cromosómica, que ocurre en el momento de la concepción. Se produce en uno de cada 700 nacimientos, en todos los grupos étnicos y el número de afectados suele ser mayor en los niños que en las niñas no se sabe con exactitud porque. Cada célula del organismo lleva en su interior el patrimonio genético que caracteriza a cada persona. Este está contenido en los cromosomas, que en número de 46 por cada célula, identifican a la especie humana. Estos se presentan pareados, de tal forma que existen 23 pares por cada célula . Cuando se gesta un nuevo ser, éste heredo dos copias de cada cromosoma: uno de la madre y otra del padre, claro que la suma de ambos dará nuevamente 46 (23+23). Sin embargo, hay veces que aparece un cromosoma extra en el par Nº21 un cromosoma supernumerario, una especie de "convidado de piedra" y es esto lo que caracteriza al SD o Trisomía 21. Allí donde debían haber 2 cromosomas, ahora hay tres. El niño nace entonces con una clara diferencia con el resto de sus semejantes, porque mientras todos tienen 46 cromosomas, él cuenta con 47. Las características físicas de estos menores ayudan al diagnóstico temprano, casi al momento del parto: diámetro cefálico reducido, ojos almendrados, dedo meñique atrofiado, dedo gordo del pie separado de los otros, orejas pequeñas. El coeficiente intelectual promedia en 50, pero además de esta menor capacidad cognitiva y de los mencionados rasgas físicos, no existen grandes diferencias en su desarrollo y adquisición de hábitos con él esto de los niños. Realidad cada día más asumida entre los padres que ven con menos temor la llegada de este hijo diferente, a quien le aprenden a descubrir enormes cualidades y valores. Unas Fotos.. Muchas Gracias a : Yahoo Por ayudar a realizar este post. Saludos Atte: Camila (Yolachiqui) Por favor si no les gusto el post no insulten solo denuncien..
¿Cuales son las principales discapacidades? Existen cinco principales tipos de discapacidad, clasificadas según los ámbitos del ser humano que afectan: * Discapacidad auditiva... * Discapacidad física.... * Discapacidad mental... * Discapacidad psíquica * Discapacidad visual Conceptos * Deficiencia: Es toda pérdida o anormalidad de una estructura o función sicológica, fisiológica o anatómica. * Discapacidad: Es toda restricción o ausencia (debido a una deficiencia) de la capacidad de realizar una actividad en la forma o dentro del margen que se considera normal para un ser humano. * Minusvalía: Es una situación desventajosa para un individuo determinado, consecuencia de una deficiencia o de una discapacidad, que limita o impide el desempeño de un papel que es normal en su caso (en función de su edad, sexo y factores sociales y culturales). Discapacidades Alzheimer Discapacidad Psíquica Lesiones Medulares Ataxia Esclerosis Lateral Amiotrófica (ELA) Osteogénesis Imperfecta Cardiopatías Esclerosis Múltiple Parálisis Cerebral Distonía Extrofia Vesical Parkinson Discapacidad Auditiva Fibrosis Quística Poliomielitis Discapacidad Mental Insuficiencia Renal Crónica Síndrome de Down Discapacidad Visual Ø Alzheimer ¿Qué es el mal de Alzheimer? Científicamente, se define como una demencia progresiva y degenerativa del cerebro. Provoca en un principio un deterioro en la memoria inmediata. Las neuronas que controlan la memoria y el pensamiento se van deteriorando, interrumpiendo el paso de mensajes entre ellas. Las células desarrollan cambios distintivos: placas seniles y neurofibrilares (degeneraciones del tejido cerebral). La corteza cerebral (principal origen de las funciones intelectuales) se atrofia y encoge. ¿Cómo se diagnostica? En la actualidad, no existe una sola prueba diagnóstica para la Enfermedad de Alzheimer. Cuando se observen los síntomas, se debe obtener una evaluación física, psiquiátrica y neurológica completa, realizada por un médico con experiencia en el diagnostico de trastornos de demencia. ¿Hay tratamiento? Esta enfermedad no tiene cura, pero tiene tratamientos que se utilizan para paliar la situación. Hay medicamentos que ayudan a desacelerar el deterioro de la acetilcolína, un producto químico necesario para la comunicación de cerebro-célula, este se agota gravemente en Alzheimer. También para el síntoma de deambulación, agitación y la alteración del sueño. El sentirse cuidado con amor, con muchísima paciencia y compresión, es la mejor medicación que podemos suministrarles. Ø Ataxia ¿Qué es la Ataxia? Es una enfermedad degenerativa de origen neurológico. Gran parte de las Ataxias son de transmisión hereditaria, ya sea de forma dominante o recesiva. Este es el motivo por el que a los afectados se les plantean serias dudas a la hora de enfrentarse a una futura paternidad. ¿Cómo se diagnostica? Los primeros síntomas frecuentemente son tropiezos o un caminar similar al de alguien ligeramente ebrio, seguido por inestabilidad e incoordinación de movimientos en los brazos y las manos. En algunos pacientes los síntomas progresan con bastante rapidez durante un periodo de 10 a 15 años, o despacio en otros pacientes durante un espacio de 20 años o más. Los afectados en el futuro necesitarán una silla de ruedas que les proporcionará independencia de movimientos. La Ataxia es una vida reducida por la enfermedad. Los afectados experimentarán dificultades para permanecer de pie, caminar, correr y mantener el equilibrio. ¿Hay tratamiento? Esta enfermedad no tiene cura. Ø Cardiopatías Comunicación interauricular. Las comunicaciones interauriculares (CIA) se producen por defectos en el tabique interauricular, lo cual hace que la sangre fluya desde la aurícula izquierda hacia la aurícula derecha. La localización del defecto en el tabique interaricular es variable, por lo que se suelen distinguir varios tipos. El defecto del tipo seno venoso se localiza en la parte alta del tabique interauricular, próximo a la entrada de la cava superior. La CIA tipo ostium secundum es la más frecuente y se localiza en la parte media del tabique interauricular. La CIA tipo ostium primum se localiza en la parte baja del tabique interauricular, inmediatamente adyacente a las válvulas aurículo-ventriculares, y es una forma de defecto del canal aurículo-ventricular. Un caso especial de cortocircuito izquierda-derecha a nivel auricular es el que se produce por la ausencia congénita del tabique interauricular, lo que da lugar a una aurícula única, que es una anomalía poco frecuente. Etiología. La causa de los defectos del tabique interaricular no es bien conocida. Lo más frecuente es que se encuentren de forma aislada, sin causa evidente ni enfermedad concomitante. En algunos casos, sin embargo, se encuentra una etiología. Así, la rubeola de la madre durante el embarazo se ha relacionado con defectos del tabique interaricular en el hijo. Diversos síndromes genéticos, como el de Ellis-van Cleveld y el de Holt-Oram muestran una incidencia aumentada de CIA, al igual que en el síndrome TAR, que cursa con trombocitopenia y agenesia o hipoplasia del radio. La aurícula única suele observarse en pacientes con síndrome de Ellis-van Cleveld o asociada a asplenia o polisplenia. En la trisomía 21 o síndrome de Down se observa una incrementada incidencia de defectos del canal aurículo-ventricular. Además, existe una forma familiar de defectos del canal aurículo-ventricular, que se transmite mediante herencia autosómica dominante. Por otro lado, la CIA tipo ostium secundum presenta 2 formas familiares, que se transmiten con una herencia autosómica dominante y que no se asocian a anomalías extracardiacas. La forma familiar más frecuente cursa con CIA tipo ostium secundum y bloqueo aurículo-ventricular. La otra forma familiar es poco frecuente y sólo muestra el defecto del tabique interaricular sin ninguna otra anomalía acompañante. Ø Discapacidad Auditiva Es la pérdida total de la audición. La prevención de la sordera es relativamente difícil debido a las numerosas causas que la provocan en los distintos períodos: prenatal, perinatal y posnatal. La causa hereditaria o genética es la más importante y desgraciadamente poco previsible. En el período perinatal, la sordera se produce por problemas de partos anormales por causa fetal o materna. Dentro de las causas posnatales de sorderas, ocupa un lugar muy importante aún la meningitis bacteriana. En segundo lugar, las otitis medias producen habitualmente un deterioro paulatino de la audición. En tercer lugar, la sordera es producida por ruidos de alta intensidad. Según el Censo de 1992, en Chile hay 59.048 personas con sordera total. La audiometría permite precisar el grado de sordera (evaluado en decibelios de pérdida), y su tipo. Así se distingue las sorderas de transmisión, que afectan al conducto auditivo externo (oído externo), el tímpano, la caja del tímpano y la cadena de huesos (oído medio), y las sorderas de percepción, que corresponden a una lesión de la cóclea o del nervio auditivo (oído interno). Ø Discapacidad Mental Según la American Association of Mental Retardation, el retraso mental alude a limitaciones sustanciales en el funcionamiento actual. Se caracteriza por un funcionamiento intelectual significativamente inferior a la media, que generalmente coexiste junto a limitaciones en dos o más de las siguientes áreas de habilidades de adaptación: comunicación, auto-cuidado, vida en el hogar, habilidades sociales, utilización de la comunidad, auto-dirección, salud y seguridad, habilidades académicas funcionales, tiempo libre y trabajo. A menudo, junto a limitaciones especificas coexisten potencialidades en otras áreas adaptadas o capacidades personales. Algunos ejemplos de deficiencias que pueden derivar en una discapacidad mental son el síndrome de Down, el síndrome de X Frágil, el síndrome de Wesn y la Fenilcetonuria. Sin embargo, existen muchas más. Ø Discapacidad Psíquica Se considera que una persona tiene discapacidad psíquica cuando presenta "trastornos en el comportamiento adaptado, previsiblemente permanentes". ¿Cuáles son las causas? La discapacidad psíquica puede ser provocada por diversos trastornos mentales, como la depresión mayor, la esquizofrenia, el trastorno bipolar; los trastornos de pánico, el trastorno esquizomorfo y el síndrome orgánico. Ø Discapacidad Visual ¿Qué es la Ceguera? Según la Organización Mundial de la Salud (OMS), es aquella visión menor de 20/400 ó 0.05, considerando siempre el mejor ojo y con la mejor corrección. Se considera que existe ceguera legal cuando la visión es menor de 20/200 ó 0.1 en el mejor ojo y con la mejor Corrección. ¿Qué es baja Visión? Es una visión insuficiente, aun con los mejores lentes Correctivos, para realizar una tarea deseada. Desde el punto de vista funcional, pueden considerarse como personas con baja visión a aquellas que poseen un resto visual suficiente para ver la luz, orientarse por ella y emplearla con propósitos funcionales. ¿Cuáles son las Causas? Aunque la ceguera puede ser provocada por algún accidente, también existen numerosas enfermedades que la desencadenan: Catarata, glaucoma, leucomas cornéales, retinopatía diabética, retinopatía del prematuro, catarata y glaucoma congénitas, atrofia óptica, distrofia retinal y retinosis pigmentaria, entre otras. ¿Se puede prevenir? Existen diversas maneras de prevenir la discapacidad visual, como el evitar accidentes del tránsito, del trabajo y enfermedades ocupacionales; atención adecuada del embarazo; detección y registro de deficiencias en los recién nacidos y el asesoramiento genético a las familias en los casos de enfermedades hereditarias. La consulta oftalmológica precoz cuando hay antecedentes en la familia también contribuye a la prevención. Ø Distonía Muscular ¿Qué es Distonía? Distonía es tanto el síntoma como el nombre de un grupo de enfermedades, precisamente denominadas distonías. El síntoma o manifestación física, consiste en permanentes contracciones involuntarias de los músculos de una o más partes del cuerpo. Esto a menudo se manifiesta en torsiones o deformaciones de esa parte del cuerpo. El trastorno es secundario a la disfunción del sistema nervioso central, probablemente en aquellas partes del cerebro llamados ganglios basales. En la de tipo primaria o distonía sin complicaciones, no hay alteración de la conciencia, de las sensaciones o de la función intelectual. La distonía puede estar acompañada de temblor, algunas veces parecido a los temblores que generalmente se observan en personas de edad, o por un temblor de tipo severo, de calidad irregular e inestable. ¿Qué hacer si se tiene un diagnóstico de Distonía? Si se considera un diagnóstico de distonía, lo primero que se debe hacer es revisar cuidadosamente el árbol genealógico de la familia, buscando personas que tengan distonía focal o generalizada. Aún el más simple "cuello torcido" (tortícolis) debe ser tomado en cuenta. Como también cualquier tipo de temblor. Esta información es muy significativa para un examen genético. En segundo lugar, se debe estudiar el tipo de terapia médica a seguir. Aquí un neurólogo con una considerable experiencia debe ser su guía. Muchos medicamentos están disponibles. Cualquier medicamento puede ser moderadamente efectivo, o medianamente efectivo, o simplemente no ser de ninguna ayuda. Sin embargo, la persistencia en probar medicamentos puede ser finalmente recompensada. Terapia con medicamentos Algunos de los medicamentos que su doctor puede considerar incluyen: Artane (trihexyphenidyl), Cogentin (benztropine), Valium (diazepam), Klonapin (clonazepam), Lioresal (baclofen), Tegretol (carbamazepine), Sinemet o Madopar (levodopa), Parlodel (bromocriptine), Symmetrel (amantadine). Ø Esclerosis Lateral Amiotrófica (ELA) ¿Qué es la ELA? La ELA es una enfermedad neuromuscular en la que las células nerviosas, las motoneuronas que controlan el movimiento de la musculatura voluntaria, gradualmente disminuyen su funcionamiento y mueren, provocando debilidad y atrofia muscular. Estas motoneuronas se localizan en el cerebro y en la médula espinal. ¿Cuál es la causa de la ELA? La causa de la ELA es desconocida. En cualquier caso, es un hecho que el conocimiento que se tiene del funcionamiento del sistema nervioso es cada vez mayor, y aumenta cada año, gracias a la utilización de herramientas más sofisticadas en el ámbito de la biología molecular, ingeniería genética y bioquímica. Todo esto nos hace albergar mayores esperanzas a la hora de pensar en un pronto descubrimiento de su origen. ¿Cómo se diagnostica la ELA? El diagnóstico es fundamentalmente clínico, es decir, no existe ninguna prueba específica que dé el diagnóstico definitivo. Después de que se haya confirmado el diagnóstico de ELA, se deben practicar numerosas pruebas de distinto tipo para descartar otras enfermedades que pueden simular la ELA. Con estos test, el estudio de la historia clínica del paciente y un detenido examen neurológico, los especialistas suelen llegar al diagnóstico definitivo. De entre las pruebas que se deben realizar para su diagnóstico, destacan una resonancia nuclear magnética, cerebral o espinal, un estudio electromiográfico de la función neuromuscular y una batería de análisis de sangre y de orina específicos. Nosotros recomendamos siempre que los pacientes tengan un segundo diagnóstico realizado por un médico con experiencia en ELA, con el fin de reducir el número de diagnósticos incorrectos. En muchas ocasiones, el diagnóstico definitivo puede tardar varios meses en producirse, aún después de realizar todos los test pertinentes y observar atentamente la evolución de los síntomas. ¿Existe algún tratamiento? Por ahora no existe ningún tratamiento probado contra la ELA Sin embargo, el reciente descubrimiento de determinados factores de crecimiento neuronal y de agentes bloqueantes del glutamato, se han mostrado prometedores en la detención de la progresión de la enfermedad, aunque no existe aún ningún fármaco que la cure. Sí existen fármacos para combatir el cortejo sintomático que acompaña a la enfermedad, como son los calambres, la espasticidad, las alteraciones en el sueño o los problemas de salivación. Existen numerosas estrategias muy eficaces para cuando aparecen las alteraciones respiratorias o cuando surgen problemas relacionados con las secreciones. Los fisioterapeutas, terapeutas ocupacionales y logopedas, son los profesionales encargados de asegurar la independencia funcional a través del ejercicio y la utilización de los equipos técnicos oportunos. Ø Esclerosis Múltiple ¿Qué es la esclerosis múltiple? La esclerosis múltiple (EM) es una enfermedad del sistema nervioso central (SNC) en la que se diferencian dos partes principales: cerebro y médula espinal. Envolviendo y protegiendo las fibras nerviosas del SNC hay un material compuesto por proteínas y grasas llamado mielina que facilita la conducción de los impulsos eléctricos entre las fibras nerviosas. En la EM la mielina se pierde en múltiples áreas dejando en ocasiones, cicatrices (esclerosis). Estas áreas lesionadas se conocen también con el nombre de placas de desmielinización. La mielina no solamente protege las fibras nerviosas sino que también facilita su función. Si la mielina se destruye o se lesiona, la habilidad de los nervios para conducir impulsos eléctricos desde y al cerebro se interrumpe y este hecho produce la aparición de síntomas. Afortunadamente la lesión de la mielina es reversible en muchas ocasiones. La EM no es ni contagiosa, ni hereditaria, ni mortal. ¿Cuáles son los síntomas de la EM? Los síntomas incluyen debilidad, hormigueo, poca coordinación, fatiga, problemas de equilibrio, alteraciones visuales, temblor, espasticidad o rigidez muscular, trastornos del habla, problemas intestinales o urinarios, andar inestable (ataxia), problemas en la función sexual, sensibilidad al calor, problemas de memoria a corto plazo y ocasionalmente problemas de juicio o razonamiento (problemas cognoscitivos). Hay que remarcar que la mayoría de personas con EM no tienen todos estos síntomas. Ø Espina Bífida Malformación sin causa Es una malformación congénita que origina diversas alteraciones en el organismo y que tiene distintos grados de afección. Espina Bífida (EB) es una de las malformaciones más graves del tubo neural, compatibles con una vida prolongada. Se da en el nacimiento por una falta de cierre o fusión de varios arcos vertebrales. El defecto se origina precozmente en el primer mes de gestación. Las causas son desconocidas, pero se cree que es la resultante de una combinación de factores genéticos y ambientales. Esta anomalía puede estar localizada en cualquier zona de la columna vertebral y dependiendo de su grado de gravedad hay tres tipos diferentes de EB: espina Bífida oculta se halla en la parte inferior de la columna y consiste en una pequeña obertura de un arco vertebral. Ø Extrofia Vesical ¿Qué es la Extrofia Vesical? La palabra extrofia se deriva de la palabra griega ekstriphein que literalmente significa vuelta o giro de dentro a fuera. La extrofia de la vejiga es una malformación de la vejiga y uretra, en la que la vejiga ha girado de adentro a fuera. La uretra, que es el canal que lleva la orina fuera del cuerpo, no se ha formado completamente. Esto se llama epispadias. En chicos, el pene tiene apariencia aplastada y podría estar levantado (alzado) hacia el abdomen. En las niñas, la uretra abierta es localizada entre un clítoris dividido y el labio menor. ¿Qué determina la magnitud de la Extrofia? La magnitud de la extrofia depende de cuan pequeña o grande es la apertura. Uno puede pensar en la extrofia como un espectro de condiciones que van desde la forma más apacible, epispadias (sólo un defecto en la uretra o apertura), a la forma más severa, extrofia cloacal (defecto en la uretra, vejiga e intestino), estando la extrofia vesical clásica (defecto en la uretra y la vejiga) en el medio de las dos anteriores. ¿Qué causa la Extrofía Vesical? La ciencia médica no puede explicar el por qué un embrión puede desarrollar extrofia vesical. No se debe a nada que el padre o la madre hizo o no hizo durante el embarazo. Por otro lado, no es simplemente una condición heredada. Ø Fibrosis Quística ¿Qué es la Fibrosis Quística? La Fibrosis Quística es una enfermedad crónica, en la mayoría de los casos degenerativa y de carácter genético (no se adquiere a lo largo de la vida) y hereditaria, trasmitida conjuntamente por ambos progenitores y que ocasiona una patología de tipo evolutivo que varía de una persona a otra. Debe negarse rotundamente que se trate de una enfermedad contagiosa, por lo que el contacto con una personas afectada no reviste peligro de contagio. Ø Insuficiencia Renal Crónica ¿Qué es la Insuficiencia Renal Crónica? Se entiende por insuficiencia renal la pérdida de la función de los dos riñones. Habitualmente, esta pérdida se produce a la vez en ambos y es importante destacar que un sólo riñón sin problemas es suficiente para mantener una función completamente normal. La insuficiencia renal puede ser aguda cuando aparece de forma brusca y normalmente tiende a recuperarse, y crónica cuando el fallo de función de los riñones se produce de forma lenta y progresiva, sin posibilidades de recuperación. Ø Lesiones Medulares Cualquier daño a la médula espinal es una lesión muy compleja. Cada lesión es diferente y puede afectar el cuerpo en varias formas diferentes. Este en un breve resumen de los cambios que suceden después de una lesión en la médula espinal. Esto dice como la médula espinal funciona y qué puede pasar con el cuerpo después de una lesión en la médula espinal. La Médula Espinal Normal Los nervios son estructuras similares a un cordón compuestos de muchas fibras nerviosas. La médula espinal tiene muchas fibras nerviosas espinales. Las fibras nerviosas transportan mensajes entre el cerebro y las diferentes partes del cuerpo. Los mensajes pueden ser relacionados con el movimiento, diciéndole a alguna parte del cuerpo que se mueva. Otras fibras nerviosas llevan mensajes de sensación o tacto desde el cuerpo hacia el cerebro, como el calor, el frío o el dolor. El cuerpo también tiene un sistema nervioso autonómico. El controla las actividades involuntarias del cuerpo como la presión sanguínea, la temperatura corporal y el sudor. Estas fibras nerviosas constituyen el sistema de comunicación del cuerpo. La médula espinal puede ser comparada a un cable telefónico. Conecta la oficina principal (el cerebro), con otras oficinas particulares (las partes del cuerpo) por medio de líneas telefónicas (las fibras nerviosas). La médula espinal es el camino que los mensajes usan para viajar entre el cerebro y las diferentes partes del cuerpo. Debido a que la médula espinal es una parte vital de nuestro sistema nervioso, está rodeado y protegido por huesos llamados vértebras. Las vértebras, o huesos de la espalda, están colocadas una arriba de la otra y se llama la columna vertebral o la columna espinal. La columna vertebral es el soporte número uno del cuerpo. La médula espinal realmente pasa por en medio de las vértebras. La médula espinal tiene aproximadamente 45 cm. de largo. Se extiende desde la base del cerebro, continúa hacia abajo de la mitad de la espalda, aproximadamente hasta la cintura. El haz de fibras nerviosas que constituyen la médula espinal por si mismo son las neuronas motoras superiores (UMN). Los nervios espinales se ramifican desde la médula espinal hacia arriba y hacia abajo de el cuello y la espalda. Estos nervios, neuronas motoras inferiores (LMN), salen entre cada vértebra y alcanzan todas las partes del cuerpo. La médula espinal termina cerca de la línea de la cintura. Desde este punto, las fibras nerviosas espinales bajas continúan hacia abajo a través del canal espinal hasta el sacro o coxis. La columna espinal está dividida en cuatro secciones o partes. La porción superior, es nombrada el área cervical, tiene siete vértebras cervicales. La sección que sigue, la dorsal, incluye el área del pecho y tiene doce vértebras dorsales. La sección baja de la espalda es nombrada el área lumbar. Hay cinco vértebras lumbares. La sección final tiene cinco vértebras sacras y es nombrada el área sacra. Los huesos en la sección sacra, en realidad están fusionados en un solo hueso. Ø Osteogénesis Imperfecta ¿Qué es la Osteogénesis Imperfecta? La osteogénesis imperfecta es una enfermedad congénita que se caracteriza porque los huesos de las personas que la sufren se rompen muy fácilmente, con frecuencia tras un traumatismo mínimo e incluso sin causa aparente. Se conocen varios tipos de la enfermedad, y su variación es muy grande de un individuo a otro. Incluso dentro del mismo tipo, puede haber personas con una mayor o menor impregnación. Por decirlo con un ejemplo práctico, algunos pacientes sufren diez fracturas a lo largo de su vida, en tanto que otros pueden llegar a tener varios cientos de ellas. ¿Cuál es la causa de la Osteogénesis Imperfecta? La osteogénesis imperfecta se debe a la insuficiente y/o defectuosa formación del colágeno del cuerpo, como consecuencia de un fallo genético. El colágeno es una proteína de los tejidos y su función en la formación de los huesos se puede comparar a la de las nervaduras metálicas en torno a las cuales se monta la estructura de hormigón de una viga. Si la nervadura no es fuerte o no existe, la pieza de hormigón no adquirirá la forma adecuada o será sumamente frágil. Ø Parálisis Cerebral ¿Qué es la Parálisis Cerebral? Es un conjunto de desórdenes cerebrales que afecta el movimiento y la coordinación muscular. Es causada por daño a una o más áreas específicas del cerebro, generalmente durante el desarrollo fetal, pero también puede producirse justo antes, durante o poco después del nacimiento, así como en la infancia. Existen diversos grados de parálisis cerebral. Además, tradicionalmente se distinguen cuatro tipos de Parálisis Cerebral: Espástica (la manifestación más común, en que los músculos permanecen tensos, parecen rígidos y los movimientos voluntarios como el caminar son difíciles; a veces pueden); Disquinética (que se caracteriza por movimiento involuntario de la cara, las manos y otras partes del cuerpo); Atáxica (que causa problemas de equilibrio y coordinación, particularmente al caminar) y Mixta (que puede combinar cualquiera de los tipos anteriores). ¿Cuáles son las causas? Puede ser producida por una serie de factores. Una causa importante es la falta de oxígeno en el cerebro durante la etapa fetal o de recién nacido, que puede deberse a una separación prematura de la placenta desde la pared del útero, mala posición natal del bebé, un parto muy largo o abrupto o interferencia de la circulación en el cordón umbilical. También puede causar Parálisis Cerebral una incompatibilidad sanguínea entre la madre y el infante o la infección de la madre con virus y otros microorganismos que atacan el sistema nervioso del infante. Una herida en el cráneo o falta de oxigenación por inmersión también son causas frecuentes, como resultado de accidentes. ¿Cuáles son las características? Dependiendo de qué áreas del cerebro tiene dañadas; una persona con Parálisis Cerebral puede presentar algunas de las siguientes características: Tensión muscular o espasmos; movimientos involuntarios; sensación y percepción anormales; limitación visual, auditiva o del habla; tercianas; retraso mental. Quienes tienen Parálisis Cerebral también pueden presentar dificultades para comer para controlar la vejiga y los esfínteres, para respirar debido a problemas posturales, escaras, o discapacidades para el aprendizaje. Ø Parkinson El hallazgo de un factor tóxico-ambiental como responsable de alteraciones parkinsonianas, ha producido un importante impulso en el ámbito de la investigación de la causa de esta enfermedad. ¿Cuáles son los síntomas? La enfermedad de Parkinson se manifiesta básicamente por la presencia de temblor, rigidez de los músculos y lentitud y dificultad para iniciar el movimiento. Estos síntomas pueden aparecer aislados o combinados, pudiendo predominar en una parte del cuerpo o bien ser más marcado un síntoma sobre los demás, de forma que hay gran variación de un enfermo a otro. En los comienzos pueden aparecer molestias muy variables, difíciles de relacionar con la enfermedad. No es raro que los pacientes acudan inicialmente al traumatólogo, aquejando dolores en las articulaciones, o al psiquiatra buscando el tratamiento de un estado depresivo. Ø Poliomietis El virus usualmente entra al cuerpo por el conducto alimentario y afecta a varias partes del sistema nervioso central. Los períodos de la incubación tienen una duración de aproximadamente entre 4 a 35 días. Síntomas tempranos incluyen fatiga, dolor de cabeza, fiebre, vómitos, estreñimiento, tiesura del cuello, ó, menos normalmente, diarrea y dolor en las extremidades. Como las células no se reemplazan, los nervios que controlan el movimiento muscular se destruyen, y la infección del poliovirus puede causar parálisis permanente. Cuando las células atacadas son en centros respiratorios, el control de la respiración, se destruye y se debe aislar al enfermo en un pulmón férrico (Respiración Artificial). Aspectos clínicos El cuadro más característico se compone de fiebre elevada de instauración brusca, dolores musculares y parálisis bastante particulares. La aparición de las parálisis es repentina y su topografía anárquica. El déficit motor afecta a cualquier músculo, de manera asimétrica y desigual. A él se asocia una disminución de los reflejos osteotendinosos y una rápida tendencia a la atrofia muscular. Un detalle importante es que en esta enfermedad no existen nunca trastornos de la sensibilidad. Ø Síndrome de Down El Síndrome de Down (SD) es un accidente genético al que cualquier persona esta expuesta. Es una alteración cromosómica, que ocurre en el momento de la concepción. Se produce en uno de cada 700 nacimientos, en todos los grupos étnicos y el número de afectados suele ser mayor en los niños que en las niñas no se sabe con exactitud porque. Cada célula del organismo lleva en su interior el patrimonio genético que caracteriza a cada persona. Este está contenido en los cromosomas, que en número de 46 por cada célula, identifican a la especie humana. Estos se presentan pareados, de tal forma que existen 23 pares por cada célula . Cuando se gesta un nuevo ser, éste heredo dos copias de cada cromosoma: uno de la madre y otra del padre, claro que la suma de ambos dará nuevamente 46 (23+23). Sin embargo, hay veces que aparece un cromosoma extra en el par Nº21 un cromosoma supernumerario, una especie de "convidado de piedra" y es esto lo que caracteriza al SD o Trisomía 21. Allí donde debían haber 2 cromosomas, ahora hay tres. El niño nace entonces con una clara diferencia con el resto de sus semejantes, porque mientras todos tienen 46 cromosomas, él cuenta con 47. Las características físicas de estos menores ayudan al diagnóstico temprano, casi al momento del parto: diámetro cefálico reducido, ojos almendrados, dedo meñique atrofiado, dedo gordo del pie separado de los otros, orejas pequeñas. El coeficiente intelectual promedia en 50, pero además de esta menor capacidad cognitiva y de los mencionados rasgas físicos, no existen grandes diferencias en su desarrollo y adquisición de hábitos con él esto de los niños. Realidad cada día más asumida entre los padres que ven con menos temor la llegada de este hijo diferente, a quien le aprenden a descubrir enormes cualidades y valores. Unas Fotos.. Muchas Gracias a : Yahoo Por ayudar a realizar este post. Saludos Atte: Camila (Yolachiqui) Por favor si no les gusto el post no insulten solo denuncien..