nelsonled
Usuario (Argentina)
Hola a todos, BIENVENIDOS A MI NUEVO POST. Hace 2 años estoy trabajando en una empresa metal mecanica, en la parte de diseño en computadora de las maquinas y herramientas que fabrica dicha empresa. Algunos de mis trabajos son: DESMALEZADORAS: NIVELADORA: PATA DE CABRA PARA BANQUINA Y TRACTOR: ROLO VIAL: ROLO TRITURADOR DE RASTROJOS ARTICULADOS: ROLO TRITURADOR: SACATRONCOS: Espero que les interese el tema y cualquier duda o consulta sobre este programa estoy a su dispocision.
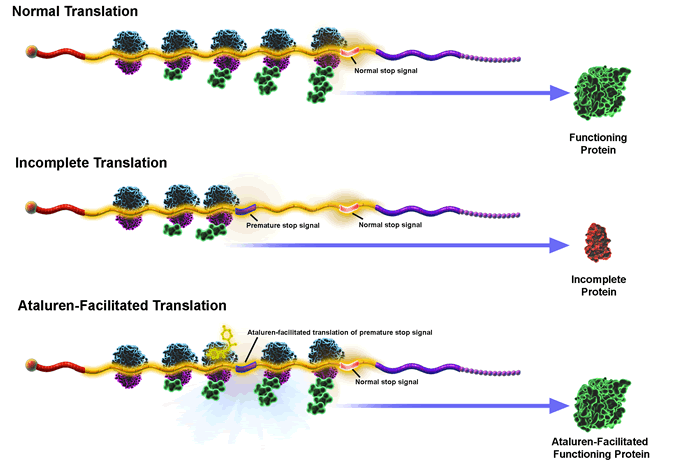
Hola a todos como andan aca les dejo un avance sobre la cura de la Distrofia muscular de duchenne. Soy de Chaco y me intereso subir este POST (el primero) porque tengo esta enfermedad y se lo que cuesta aceptarla y con esta informacion sepan que hay esperanza. SALUDOS PARA TODOS NELSON HAY DOS TRATAMIENTOS DE LOS CUALES SE ESTAN HACIENDO PRUEBAS ESTE ESUNO: AVI BioPharma Anuncia el Tratamiento del Primer Paciente en Prueba Clínica Sistémica de AVI-4658 para el Tratamiento de la Distrofia Muscular de Duchenne Portland, OR - 19 de febrero de 2009 - AVI BioPharma, Inc., un desarrollador de fármacos basados en ARN, anunció hoy el tratamiento del primer paciente en un ensayo clínico de evaluación de la entrega sistémica de AVI-4658 para el tratamiento de distrofia muscular Duchenne (DMD). "Estamos muy contentos de comenzar la evaluación sistémica de nuestro fármaco de omisión de exón (exon skipping) - AVI-4658 - para el tratamiento de la DMD," dijo Stephen Shrewsbury, MD, Director Médico Oficial y Vice Presidente Senior, de Asuntos Clínicos y Regulatorios de AVI BioPharma. "Creemos que esta prueba se basará en gran medida en los datos generados por la reciente prueba exitosa de evaluación de la administración intramuscular del mismo fármaco en niños con DMD." La prueba enroló a 16 niños con DMD ambulatorios y evaluara inicialmente dosis múltiples por vía intravenosa de AVI-4658 de entre 0.5 – 4.0 mg/kg. Esta es una prueba abierta de seguridad de 12 semanas, que incluye medición de la eficacia del medicamento y la farmacocinética. El estudio clínico inició en Londres en el Reino Unido en instalaciones del Instituto de Salud Infantil UCL / Hospital NHS Trust Gran Ormond Street por miembros del Consorcio MDEX, dirigido por el profesor Francesco Muntoni, y en breve empezaran a reclutar pacientes en Newcastle Upon Tyne. AVI BioPharma es el patrocinador de la prueba y el profesor Muntoni ha sido galardonado con el apoyo financiero de 1.3 millones de dólares del Consejo de Investigación Médica del Reino Unido para compensar algunos de los costos clínicos del juicio. En enero de 2009, AVI anunció los resultados de una Fase 1 de prueba evaluando la administración intramuscular de AVI-4658 para el tratamiento de la DMD, también realizada en colaboración con el Consorcio MDEX. Datos de la biopsia mostraron que la inyección del fármaco en los músculos de una serie de pacientes con DMD, exitosamente indujo la producción de distrofina de forma sensible por una dosis. Además, el fármaco fue bien tolerado, sin eventos adversos relacionados con el fármaco. A la empresa se le ha concedido una designación de medicamento huérfano para AVI-4658 por la Administración de Fármacos y Alimentos de EUA en noviembre de 2007 y por la Agencia Europea de Medicamentos en diciembre de 2008. La DMD es una enfermedad incurable de pérdida muscular asociada a errores en el gen que codifica la distrofina, una proteína que desempeña un importante papel estructural en la función de la fibra muscular. El AVI-4658 está diseñado para omitir el exón 51 del gen de la distrofina, lo que permite la restauración del marco de lectura en la secuencia del ARNm. Basándose en su investigación pre-clínica y los resultados de la Fase 1 de prueba, AVI cree que, al omitirse este exón, una forma truncada pero funcional de la proteína distrofina es producida para mejorar el proceso de la enfermedad, potencialmente prolongar y mejorar la calidad de vida en estos pacientes. Acerca de la Distrofia Muscular Duchenne (DMD) La DMD es uno de los trastornos genéticos comunes más mortales que afectan a niños de todo el mundo. Aproximadamente uno de cada 3500 niños en todo el mundo se ve afectado con distrofia muscular Duchenne, con 20.000 nuevos casos cada año. Se trata de una devastadora e incurable enfermedad de pérdida muscular, asociada con errores innatos en el gen que codifica la distrofina, una proteína que desempeña un importante papel estructural en la función de la fibra muscular. Los síntomas suelen aparecer en los niños antes de los 6 años. Debilidad muscular progresiva de las piernas y la pelvis eventualmente se extiende a los brazos, el cuello, y músculos respiratorios. Cerca de los 12 años de edad, los pacientes están confinados a una silla de ruedas. Finalmente, la condición progresa a la parálisis completa con el aumento de dificultad en la respiración. La condición es terminal y deceso normalmente ocurre antes de la edad de 30 años. En la actualidad no hay cura para la DMD, pero por primera vez en décadas, existen terapias prometedoras en movimiento o en desarrollo clínico. Acerca de AVI BioPharma AVI BioPharma se centra en el descubrimiento y desarrollo de fármacos basados en ARN, utilizando derivados de su propiedad de la química en antisentido (oligómeros fosforodiamidos morfolinos modificados o PMOs) que puede aplicarse a una amplia gama de enfermedades y trastornos genéticos a través de distintos mecanismos de acción. A diferencia de otros enfoques terapéuticos basados en ARN, la tecnología antisentido de AVI se ha utilizado para dirigirse más directamente tanto al ARN mensajero (ARNm), como sus precursores (ARNpre-m), que permite aumentar y disminuir la actividad de determinados genes y proteínas. Los programas de fármacos basados en ARN de AVI se están evaluando para el tratamiento de la distrofia muscular Duchenne, así como para el tratamiento de la restenosis cardiovascular a través de nuestro socio Global Therapeutics, una compañía de Cook Group. Los programas antivirales de AVI han demostrado resultados prometedores en infección por el virus Zaire, Ébola y Marburgo Musoke y pueden ser aplicables a otros objetivos, tales como virus HCV o virus del dengue. Para más información, visite www.avibio.com. Acerca del Consorcio MDEX El consorcio MDEX liderado por el profesor Muntoni, es una empresa multidisciplinaria para promover la investigación traslacional en distrofias musculares, y está formado por los grupos clínicos del Profesor Francesco Muntoni (Colegio Imperial de Londres e Instituto de Salud Infantil UCL) y la Profesora Kate Bushby y el Profesor Volker Straub (Universidad Newcastle), y científicos del Colegio Imperial de Londres (Profesor Dominic Wells), Instituto de Salud Infantil UCL (Dr. Jennifer Morgan), Royal Holloway, Universidad de Londres (Profesor George Dickson y el Dr. Ian Graham), la Universidad de Oxford (Dr. Matthew Wood) y la Universidad de Australia Occidental (Prof. Steve Wilton). Además, los grupos de caridad Muscular Dystrophy Campaign (MDC), Action Duchenne y Duchenne Family Support Group también participan en el Consorcio. Para más información, visite www.mdex.org.uk. ESTA ES EL OTRO TRATAMIENTO: La droga es Ataluren (antes PTC124 ™) es la primera investigación de nuevos medicamentos diseñados para permitir la formación de un funcionamiento de proteínas en los pacientes con trastornos genéticos debido a una mutación sin sentido. Una mutación sin sentido es una alteración en el código genético que prematuramente se detiene la síntesis de una proteína esencial. Ataluren Actualmente se está investigando para su uso en pacientes con mutaciones sin sentido la distrofia muscular de Duchenne (nmDMD) y sin sentido de mutación de fibrosis quística (nmCF). Figura 1. Traducción de ARNm en una proteína: la comparación de la traducción normal, la terminación prematura de la traducción, y el tratamiento con ataluren funcionamiento de inducir la síntesis de proteínas. Mecanismo de acción En individuos sanos, los ribosomas traducen el código de información en el ARNm en proteínas, hasta llegar a una señal de parada normal en el ARNm, momento en el que se detiene el ribosoma adecuadamente la traducción y el funcionamiento de la proteína es el resultados. Las mutaciones sin sentido, sin embargo, crean una señal de parada prematura en el ARNm. Esta señal de parada prematura causa que los ribosomas pongan fin a la traducción antes de que se genera el funcionamiento de proteínas, la creación reducida, de una proteína disfuncional. La enfermedad resultante es determinado por las proteínas que no puede expresarse en su totalidad y ya no es funcional (por ejemplo, la proteína distrofina nmDMD o en la proteína CFTR en nmCF). Ataluren está diseñado para permitir los ribosomas al ignorar la señal de parada prematura y continuar la traducción del ARNm, resulta en un funcionamiento de la formación de proteínas. Ataluren no causa los ribosomas a leer a través de la señal de parada normales. Ataluren, vía oral, tiene el potencial para hacer frente a la causa subyacente de la enfermedad por anular la señal de parada prematura, lo que permite la síntesis de una proteína de funcionamiento. Ataluren no altera el código genético del paciente o introduce material genético en el cuerpo. Los Institutos Nacionales de Salud (NIH) de la Oficina de Enfermedades Raras calcula que las enfermedades raras afectan a 25 millones de personas en los EE.UU. y que la mayoría de estas personas tienen trastornos genéticos. En más de 2.400 trastornos genéticos, una mutación causa la enfermedad en un promedio de 5 a 15% de los pacientes. Además de la mutación sin sentido en la distrofia muscular de Duchenne (nmDMD) y sin sentido de mutación de fibrosis quística (nmCF), estos trastornos genéticos incluyen una gama de enfermedades graves a través de múltiples áreas terapéuticas, incluyendo, atrofia muscular espinal, hemofilia, desórdenes de almacenamiento lisosomal, y algunas formas de cáncer. Pruebas genéticas Ataluren tiene el potencial para el tratamiento de cualquier enfermedad genética causada por una mutación sin sentido. Aunque la participación actual de los ensayos clínicos sólo absurdo y sin sentido DMD mutación la mutación de FQ, en el futuro juicio se prevé en otros trastornos genéticos causados por una mutación sin sentido. Para determinar si un trastorno genético que es causado por una mutación sin sentido, los pacientes requieren pruebas genéticas. Las pruebas genéticas se realiza mediante una simple prueba de sangre que es ordenado por un médico que trabaja en conjunto con un laboratorio genético. Los laboratorios que realicen pruebas genéticas varían en función del trastorno y la ubicación. El sitio web financiado por los NIH, www.genetest.org proporciona una lista de los laboratorios y la información de contacto. Ensayos clínicos en curso Ataluren ha demostrado prueba del concepto en la Fase 2 de ensayos clínicos en el sentido de mutación DMD (nmDMD) y sin sentido de mutación de FQ (nmCF). PTC ha iniciado el registro de estudios clínicos dirigidos a entender si ataluren cómo pueden mejorar los pacientes con nmDMD y nmCF sentir y la función y para determinar si el medicamento se puede administrar con seguridad durante un largo período. • nmDMD: PTC ha inscrito un estudio clínico de fase 2 ter de ataluren en nmDMD / DMO. El estudio es un multi-centro, aleatorio, doble ciego, controlado con placebo para determinar si puede mejorar ataluren poca actividad, la función muscular y la fuerza muscular. Se está llevando a cabo en 37 centros de todo el mundo, con aproximadamente 165 pacientes. Más información está disponible en www.clincialtrials.gov (búsqueda en "PTC124" • nmCF: PTC tiene previsto iniciar a más largo plazo, estudio clínico de nmCF en 2009. El estudio será un multi-centro, aleatorio, doble ciego, controlado con placebo. Terminado de Ensayos Clínicos • 2a fase de datos sin sentido mutación DMD (nmDMD): Los datos de la Fase 2 de ensayos clínicos en pacientes pediátricos ataluren con nmDMD muestran que la administración de ataluren está asociado con la producción de distrofina funcional. Ataluren tratamiento también se ha asociado con reducciones estadísticamente significativas en la pérdida de músculo derivados creatina quinasa en la sangre. • 2a fase de datos sin sentido de mutación de FQ (nmCF): Los datos de ensayos clínicos de fase 2a de ataluren en pacientes adultos y pediátricos con nmCF muestran que la administración de ataluren resultados en la producción de CFTR funcional y mejoras estadísticamente significativas en función de canal de cloruro CFTR en las vías respiratorias. Ataluren el tratamiento se asoció con la reducción de la frecuencia de la tos y las mejoras en las pruebas de función pulmonar. • Eventos adversos y el perfil de seguridad: En todos los estudios clínicos hasta la fecha, incluida la fase 1 por voluntarios sanos estudios, ataluren ha sido generalmente bien tolerado. En la Fase 2 bis estudios, los eventos adversos han sido en gran medida con antecedentes y síntomas usualmente han sido leves. Relativa a los resultados adversos no se han determinado sobre la base de los exámenes físicos, signos vitales mediciones, ECG, o estudios de laboratorio. En consonancia con este perfil de seguridad, significa que se ha> 90% en todos los estudios. Donaciones El desarrollo de ataluren también ha sido apoyada por subvenciones de: • Fundación de Fibrosis Quística • Asociación de Distrofia Muscular • Proyecto Padres Distrofia Muscular • Oficina del FDA (food and drugs administration) • Centro Nacional de Investigación de Recursos La FDA ha concedido PTC124 (ataluren) Subparte E designación para acelerar el desarrollo, evaluación y comercialización y ha concedido de Medicamentos Huérfanos de designaciones para el tratamiento de la FQ y la DMD debido a mutaciones sin sentido. PTC124 (ataluren) también se ha concedido la condición de medicamentos huérfanos para el tratamiento de la FQ y la DMD por la Comisión Europea. Asociación con Genzyme PTC Therapeutics, Inc. y Genzyme Corporation, una colaboración exclusiva para desarrollar y comercializar ataluren. PTC ataluren se comercializan en los Estados Unidos y Canadá y Genzyme se ataluren comercializar en todos los demás países BUENO ESPERO QUE LES INTERESE ESTA INFORMACION ESPERO QUE DEJEN SUS COMENTARIOS DE TODOS MODOS ES UN PLACER CLABORAR CON USTEDES NELSON

BIENVENIDOS A MI NUEVO POST....- ESPERO QUE LES GUSTE Y PUEDAN APRENDER A MANEJAR ESTE EXELENTE PROGRMA DE DISEÑO INDUSTRIAL.................- NO PUEDO PONER LINKS DE DESCARGA, SI NECESITAN QUE LES PASE LOS TUTORIALES EN FORMATO DE TEXTO QUE FUI RECOPILANDO MANDEN MENSAJE PRIVADO O COMENTARIO Y SE LOS MANDO ... ESTOS SON LOS CANALES Y VIDEOS DE YOU TUBE MAS INTERESANTES QUE ENCONTRE PARA QUE PUEDAN EMPEZAR, Y TAMBIEN PARA REALIZAR PROYECTOS A GRAN ESCALA Diseño Mecanico para Ti. http://www.youtube.com/user/dmnamq SolidWorksTutoriales http://www.youtube.com/user/SolidWorksTutoriales SolidWorks http://www.youtube.com/user/solidworks SolidWorks Tutorial http://www.youtube.com/channel/UCtwaWPOXEBysZLh1rrPzwFw ESPERO QUE LES HALLA SIDO DE MUCHA AYUDA EL POST...................- PARA DESCARGAR EL PROGRAMA GOOGLEEENLO JEJE O ME AVISAN Y LES AYUDO A CONSEGUIRLO........ Cualquier pregunta que tengan no duden en hacerla......
Estado actual de aprobacion de tratamientos para DMD 8 de Julio de 2015 NUEVOS TRATAMIENTOS PARA DISTROFIA MUSCULAR DE DUCHENNE (DMD). ESTADO ACTUAL DE SU APROBACION. Ante la confusion surgida de versiones sobre la inminente comercializacion de nuevas drogas de investigacion para la Distrofia Muscular de Duchenne, la Asociacion Distrofia Muscular desea dejar en claro cual es el estado de las mismas. DRISAPERSEN (Salto de Exon 51)220px-Biomarin-logo El medicamento o nueva droga denominada "Drisapersen", producto de investigacion de la compañia Prosensa, actualmente bajo licencia de la empresa BIOMARIN es mas conocida como la droga capaz de inducir el salto de Exon 51 en la DMD lo que llevaria a una correccion parcial de la expresion de la proteina Distrofina en las celulas musculares. Esta droga estaria indicada y podria ofrecer potencialmente algun beneficio terapeutico en aquellos pacientes DMD (no mas del 13% del total de los DMD) en los que el tipo de mutacion genetica hace compatible al paciente. ESTADO ACTUAL Recientemente la empresa biofarmaceutica BIOMARIN libero dos comunicados de prensa fechadas el 25 y el 29 de Junio de 2105 anunciando que tanto la EMA (Agencia Europea de Medicamentos) como la FDA (Food and Drug Administration) que son Agencias u Organismos que regulan la aprobacion de farmacos en la Union Europea (UE) y en los Estados Unidos han aceptado iniciar el proceso de revision de la droga Drisapersen. La empresa BIOMARIN aspira a que dicha droga sea autorizada por ambas agencias para su comercializacion en la Union Europea y los EEUU. Es importante aclarar que para que un medicamento de investigacion pueda ser comercializado debe ser autorizado por estas agencias (EMA en la UE o FDA en los EEUU). En nuestro pais la autorizacion para la comercializacion debe provenir del ANMAT. El proceso de aprobacion en EMA o FDA involucra un primer paso (similar para ambas agencias) en el que se le exige a la empresa solicitante (en este caso BIOMARIN) presentar toda la documentacion y evidencia que le sea solicitada para el analisis cuidadoso y muy meticuloso de estos datos. Este proceso suele ser complejo e involucra para la empresa una serie de pasos y aportes de todo tipo de documentacion que debe satisfacer todos los requisitos que solicita la Agencia. Para las llamadas enfermedades huerfanas como DMD existen algunos mecanismos que facilitan la presentacion. Este primer paso es el que se ha cumplido y resta esperar la respuesta / veredicto de ambas Agencias. La EMA anuncio que en la primera mitad del 2016 daria su respuesta a la solicitud. La FDA anuncio que su veredicto estara en Diciembre del 2015. Mientras tanto debemos aguardar y este nuevo farmaco sigue solo siendo utilizado en el marco de los protocolos de investigacion medico-cientifica. PTC TRANSLARNA o ATALUREN La droga conocida como PTC 124 , luego Ataluren y ahora Translarna es una molecula disenada para pacientes con mutaciones puntuales especificas (a diferencia del Drisapersen) que surgen de los estudios de ADN. Este tipo de mutaciones (llamadas sin sentido) afecta un porcentaje de aproximadamente 10 a 15% del total de pacientes DMD de los cuales el 50% calificarian para recibir beneficios de la medicacion. ESTADO ACTUAL La empresa PTC logro en Julio del año 2014 la aprobacion condicional para la comercializacion de la droga Translarna tambien conocida como Ataluren o PTC 124. En este caso el periodo de revision fue superado en forma exitosa y la EMA (no aun la FDA) autorizo de modo condicional (es decir que el pedido debe ser renovado) su venta comercial en los paises de la UE. ADM se encuentra haciendo las gestiones necesarias para que las personas con DMD con las caracteristicas geneticas arriba explicadas puedan acceder a la medicacion bajo la modalidad de "uso compasivo" prevista por nuestra legislacion y normativas del ANMAT. Celulas Madre, entre la estafa y la ciencia Esta modalidad habilita a la importacion del farmaco sobre la base de paciente por paciente con la debida orden medica. Celulas Madre, entre la estafa y la ciencia Buenos Aires 12-06-2013 El descubrimiento de las celulas madre ha sido un enorme avance en la ciencia medica. La posibilidad de su utilizacion como forma de tratamiento, ya sea de enfermedades neurologicas o de otro tipo, ha despertado particular interes no solo en el mundo de la ciencia si no entre los pacientes y el publico general. La prensa se ha encargado de divulgar parte de las novedades aunque con informacion no siempre acertada y que la poblacion general no puede procesar adecuadamente. Enfermedades tales como el Parkinson, Alzheimer, Esclerosis Multiple y tambien Esclerosis Lateral Amiotrofica, Distrofias Musculares y otras Enfermedades Neuromusculares, son el objeto de intensa investigacion en este terreno. Es evidente que, ante la carencia de tratamientos efectivos para estas dolencias asociadas al deterioro enorme que producen tanto en funciones intelectuales como en funciones motoras, muchos bien intencionados y otros no tanto se ocuparon de alentar expectativas. Distintos tipos de celulas madre se han probado en modelos animales de enfermedades y, en algunos casos, la respuesta ha sido alentadora y ya se han comenzado algunas pruebas muy controladas en humanos. El Sistema Nervioso, por sus particularidades, enfrenta desafios propios que lo distinguen de otros tejidos tales como su complejidad y conectividad, su metabolismo, la existencia de una barrera hemato-encefalica. Esta es una barrera que aisla el cerebro y la medula espinal de la sangre, lo que dificulta el pasaje de las celulas madre al tejido nervioso y esto se suma a la multiplicidad de celulas, que contiene el tejido nervioso (no solo neuronas, tambien celulas gliales). Mas aun muchas enfermedades a las que se intenta combatir son de origen genetico por lo que, de tratarse de celulas madre linfaticas del propio individuo o de donantes familiares, deberian primero ser sometidas a tratamientos que modifiquen sus genes (doble desafio). Hasta que estas terapias sean una realidad, y muy especialmente en Enfermedades Neuromusculares, deberan resolverse aun muchos problemas tales como la eleccion del tipo de celula madre a utilizar, la forma de llegar al tejido nervioso o muscular, al segmento particular del sistema nervioso que se haya involucrado en la enfermedad en cuestion, la eleccion del modelo experimental (en el que se hagan las pruebas y experimentos) y muchisimos otros que seria imposible enumerar. Creemos que la terapia con celulas madre sera probablemente en un futuro una realidad y todos tenemos grandes esperanzas en ella aunque aun debamos esperar. Mientras tanto digamos que todo esta area se encuentra en una fase de investigacion y por el momento no existe aplicacion clinica alguna para ninguna enfermedad neurologica ni neuromuscular. Digamoslo de otro modo: Hasta hoy NO hay NINGUN paciente en el mundo que se haya curado NI beneficiado con tratamiento con celulas madre. Por otro lado debe entenderse que cualquier tratamiento de este tipo es EXPERIMENTAL y como tal debe estar regulado, vigilado y permitido por la autoridad sanitaria local (ANMAT, INCUCAI en nuestro pais, FDA en EEUU, EMEA en Europa, etc.) y tampoco esta exento de significativos riesgos. Sin embargo, esta linea de investigacion es muy promisoria y la ciencia tiene fundadas esperanzas en que el exito corone tantos recursos invertidos. Tan, o mas importante, es mencionar que de acuerdo a normas internacionales (Acuerdo de Helsinski y otros) el sujeto de la investigacion (es decir, el paciente) no debe abonar ni un solo centavo por el costo de la investigacion (tampoco su cobertura medica). Llama la atencion el crecimiento de la oferta de individuos inescrupulosos, tanto del ambito nacional como del extranjero, que seducen a pacientes desesperanzados y portadores de enfermedades neurologicas con supuestas promesas de mejoria o curacion que constituyen verdaderas estafas no solo economicas sino morales. Quienes colaboran economicamente por razones humanitarias se transforman en victimas de la estafa y de alguna manera en complices de actos que deberian estar penados por la ley. Por ultimo, indigna el silencio oficial, indigna la prensa que promueve y se hace eco de campañas sin siquiera consultar o recabar informacion en las fuentes cientificas e indigna el silencio complice de las sociedades cientificas que callando no hacen otra cosa que alentar a los estafadores de turno y estimulando el pensamiento magico de la ignorancia a la que deberia ilustrar. Dr. Alberto L. Dubrovsky Vicepresidente y Director Medico de ADM Profesor Titular de Neurociencias "“ Universidad Favaloro Profesor Adjunto de Neurologia "“ Universidad de Bs As Jefe del Departamento de Neurologia y Unidad de Enfermedades Neuromusculares Instituto de Neurociencias Fundacion Favaloro CureDuchenne junto a Prosensa 14 de Agosto de 2014 CureDuchenne colaborara con Prosensa para Mejorar el Acceso a Medicamentos experimentales para la distrofia muscular de pacientes Duchenne CureDuchenne, un grupo de defensa de los pacientes de distrofia muscular de Duchenne, ha entrado en una nueva colaboracion con Prosensa para mejorar el acceso a medicamentos experimentales para los pacientes con distrofia muscular de Duchenne. CureDuchenne proporcionara a Prosensa US $ 7 millones en fondos para ayudar en el avance de los ensayos clinicos para una variedad de farmacos para Duchenne . El financiamiento le permitira a Prosensa: Continuar los estudios de dosificacion para drisapersen en sus ensayos de fase II, tanto en America del Norte y Europa. Este medicamento podria ayudar a aproximadamente el 13% de todas las personas con distrofia muscular de Duchenne. Mover a la fase II de los ensayos para PRO044 en Europa y un ensayo de fase III controlado con placebo en los EE.UU.. Este medicamento podria apuntar a alrededor del 6% de todas las personas con Duchenne. Apoyar la investigacion y el desarrollo de otras tecnologias de drogas de salto de exon similares. La enfermedad, que solo afecta a los ninos, causa degeneracion muscular que finalmente conduce a una incapacidad para caminar y la muerte, por lo general antes de los 30 anios . La degeneracion muscular se produce debido a la falta de distrofina, una proteina que es esencial en el mantenimiento de las celulas de las fibras musculares sanas . Mediante la tecnica de Salto de Exon se pueden inducir la produccion de distrofina mediante la alteracion del marco de lectura durante la transcripcion de ADN. Espero que les sirva las novedades cualquier cosa comenten o manden mensaje privado. Hasta la Próxima